Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
107 Cards in this Set
- Front
- Back
|
Gene
|
Segment of DNA that encodes for a specific protein and certain RNAs
|
|
|
Locus
|
Region on a chromosome that contains a specific gene
|
|
|
Alleles
|
Alternative forms of a gene that differ in DNA sequens or in phenotypic consequences.
|
|
|
Chromosome
|
Structure that carries genes
|
|
|
Homologous chromosomes
|
Chromosomes that pair during meiosis, inherited from each parent. 2 copies of nearly identical chromosomes from each parent.
|
|
|
Sister chromatids
|
2 identical duplicated chromosomes (from one parent) joined by a single centromere.
|
|
|
Polidy
|
Amount of homologous chromosomes.
|
|
|
Haploid
|
1 copy of each chromosome (23 in humans)
|
|
|
Diploid
|
2 copies of each chromosome (46 in humans)
|
|
|
Aneuploid
|
Any number of chromosomes that is not an exact multiple of the haploid number
|
|
|
Monosomy/Trisomy
|
Result of non-disjunction during meiosis/mitosis resulting in absence or addition of an extra chromosome.
Usually only small chromosomes, larger ones result in death. |
|
|
Somatic cell
|
Cells that do not pass on genetic info.
|
|
|
Germ line cells
|
Cells that pass on genetic info
|
|
|
Mitosis vs Meiosis
|
Meiosis - haploid gamete cells, pairing of homologous chromosomes.
Mitosis - normal cell division, no pairing of homologous chromosomes |
|
|
Synapsis
|
Proteins bind homologous pairs of sister chromatids together, occurs in prophase 1.
|
|
|
Crossing Over
|
Recombinase expressed, genetic recombination during meiosis. 0 - 2 times on a chromosome.
|
|
|
Spermatogeneis
|
Symetrical division - all 4 turn into gametes.
Mitosis occurs throughout life and induced at puberty. ⬆Paternal Age = ⬆Point mutations |
|
|
Oogenesis
|
Asymetrical division - only 1 turns into gamete.
Mitosis occurs in fetus, all precursors are arrested in prophase I, mitosis initiated during puberty. ⬆Maternal age = ⬆Chromosomal disorders |
|
|
Promotor Sequences
|
Control levels of expression of mRNA which controls expression of protein. If mutated?
|
|
|
Coding Sequences
|
Exons, specify the sequence of amino acid in proteins, codons can code for more than 1 aa.
|
|
|
Intervening Sequences
|
Interons, interrupt coding sequences and are spliced out of mature mRNA
|
|
|
Structural Regions
|
Untranscribed regions 5' and 3' untranslated regions in mRNA
|
|
|
Splicing
|
Must have consensus sequence at splice junctions (border between interons/exons).
|
|
|
Nonsense Mut.
|
Insert stop codon, resulting in shorter protein (no fxn) or not expressed.
|
|
|
Missense Mut.
|
Changes codon specificity to new aa, expressed but may be a different fxn.
|
|
|
Frameshift Mut.
|
Small deletion or insertion that changes all subsequent codons.
Results in active and inactive proteins, just because it's active does not mean it is not detrimental. |
|
|
Gain of Function Mut.
|
Novel protein product of or over-expression/miss-expression of a normal protein, usually dominant disorders
|
|
|
Dominant Negative Mut.
|
Mutant protein inhibits activity of normal protein.
|
|
|
Lethal Mut.
|
Occurs in domains of proteins that inhibits activity of normal protein.
If dominant mutation is lethal or reproductively lethal, it can't be passed on. These mutations are random new mutations. |
|
|
Silent Mut.
|
Occur in non-essential regions (interons, 5'/3' regions, non-transcribed/non-promotor) or it occurs in redundant base in codon.
Makes a conservative subsititution (similar aa or non-essential region of protein.). Most mutations are not disease causing. |
|
|
Karyotyping
|
Lymphocytes grown in culture and arrested in metaphase, lysed and chromosomes collected, chromosomes via banding pattern on long (q) and short (p) arms and centromeric position.
Detects: Changes in chromosomes number (trisomy, monosomy), as well as large deletions, inversions, duplications and translocation. Very slow. 7p22.2 (chromosome 7, short p, band 22.2) |
|
|
Mosaicism
|
Presence of cells that have different genotype caused by high amount of chromosomal rearrangement.
Ex: Milder form of kleinfelter's syndrome 46/47 XY/XXY where some pt. cell have XY and some have XXY. |
|
|
FISH
|
Fluorescence in situ hybridization.
Specific fluorescent labeled nucleotide probes mark where chromosomes are. Quicker than regular karyotyping. Two preps are used (metaphase nuclei - determines chromosome structure and interphase nuclei - determines number). Able to look for mosaicism |
|
|
Locus Specific Probe
|
Probe in FISH.
Labels specific gene or region. Used to detect microdeletions. |
|
|
Centromeric Probe
|
Probe in FISH.
Labels specific pair of chromosomes at their centromere. |
|
|
Chromosome Painting Probe
|
Probe in FISH.
Labels a specific chromosome across entire length. Used to detect trisomies, translocations and large duplications. |
|
|
Spectral Karyotyping
|
Every chromosome is probed, usually ordered when lots of chromosomal recombination is suspected (cancer = ⬆recombinase).
|
|
|
Microarray SNP Testing
|
Chop up chromosomes, put DNA in wells w/probes. Indicated after normal karyotype.
Detects: # of chromosomes, deletion/duplication, and unbalanced translocation unable to detect balanced translocations (chopped up chromosomes) and low-level mosaicism. Quick, can determine imbalances across the entire genome and high resolution. |
|
|
Chromosomal Non-Disjunction
|
Occurs during gametogenesis, affects hundreds of genes and is random.
Can occur during: 1st meiotic division - gametes with copies of 2 diff. maternal chromosomes 2nd meiotic division - gametes with 2 sister chromatids. |
|
|
Duplications
|
Part of chromosome copied and inserted into same/different chromosome.
Unbalanced. |
|
|
Inversions
|
Section of chromosome is flipped. Can cause problems w/ segregation at gametogenesis.
Balanced. |
|
|
Insertions
|
Section of chromosome is inserted into another chromosome. Can cause problems w/ segregation at gametogenesis.
Balanced. |
|
|
Isochromosomes
|
Abnormal centromeric division, 2 long arms, 2 short arms. Problems w/ segregation at gametogenesis.
Balanced. |
|
|
Reciprocal Translocations
|
Parts of chromosomes are exchanged.
Problems w/ segregation at gametogenesis. Balanced - Indvid. are often normal/carriers (two copies of each gene). Leads to increased incidence of non-disj. in next generation. |
|
|
Robertsonian Translocations
|
Centric fusion of two chromosomes, likely to happen w/ acrocentric chromosomes.
No lost coding sequences, carrier unaffected, problems w/ segregation at gametogenesis. Leads to increased incidence of non-disj. in next generation. |
|
|
Balanced vs Unbalanced Rearrangements
|
Balanced - same amt. of genetic units brought over.
Unbalanced - not same amt. of genetic units brought over. Reproductively lethal, and caused by: spontaneous mutations (recurrence risk elevated but low) unbalanced inheritance from balanced rearrangements (recurrence risk is signif. elevated) |
|
|
Principle of Segregation
|
Genes (homologous chromo) occur in paris of individ. only 1 of each chromo is transmitted to offspring from the parent
|
|
|
Principle of Indep Assort
|
Genes at different loci are transmitted independently (True for chromosomes and for distant genes on the same chromosome, but not for close genes (linkage).
|
|
|
Co-dominance
|
Heterozygotes have different phenotype than homozygotes.
|
|
|
What makes Dom. vs Reces.
|
Consequences of mutations on protein expression. Dosage sensitivity of trait - dependence of the trait on protein expression levels/activity.
|
|
|
Consequences of Mutation
|
Loss of fxn - dom or reces. (1 mut = lose 1/2 activity, 2 mut = lose all act.).
Gain of fxn - dom because of new activity. Dom. neg. - dom. because it eliminates almost all protein activity. |
|
|
Dosage Sensitivity of Trait
|
Haploinsufficinency - 1/2 protein expression/activity is insufficient (one normal allele). Dominant.
Haplosufficiency - 1/2 protein expression/activity is sufficient (one normal allele). Recessive |
|
|
Haplosuffciency
|
One allele is enough for compensation of the mutation, usually recessive disorders
|
|
|
Haploinsuffcincy
|
One normal allele is not enough for compensation of the mutation usually dominant disorders
|
|
|
Risk of Occurrence
|
Probability that a couple will have an affected child.
Depends on population frequencies of the disorder and the rate of spont. mut. |
|
|
Risk of Recurrence
|
Probability that a genetic disorder in a family will recur in another member of in future generations.
Depends on mode of inheritence. Risk of recurrence is higher than the risk of occurrence |
|
|
Probability, Multip Rule, Add Rule
|

|
|
|
Hardy-Weinburg
|

Dom. - disease freq. approx. equal to heterozygote freq (2q; homozygotes are rare).
Reces. - disease freq.=allele freq squared (q² ). X-linked reces.- freq. of affected males = allele freq (q), and freq. of carrier females is twice the freq.of affected males (2q). X-linked dom.- freq. of affected females is twice the freq. of affected males (2q vs q) |
|
|
Autosomal Dominant
|
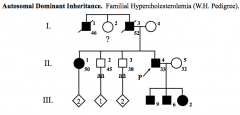
Vertical pattern of inheritance.
Father-to-son transmission of disease gene is possible. Equal number of affected males and females. Most common mating: affected heterozygote and normal homozygote. Disease freq = 2q → (2)(Frequency)(Chance of getting either allele i.e. A or a) |
|
|
Autosomal Dominant Probability
|
Problems: Punnett Square. (Mother risk)(Father risk)(Chance of getting allele)
|
|
|
Autosomal Recessive
|
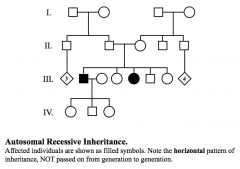
Horizontal pattern of inheritance.
Co-sanguinity is sometimes seen. Equal number of affected males and females. Most common mating is between two heterozygotes. Disease freq = q² Carrier freq. = 2q (solve q) |
|
|
Autosomal Dominant Probability
|
Problems: Punnett Square. (Mother risk)(Father risk)(Chance of getting allele)
|
|
|
2/3 Rule
|
If no affected people in sibship and know that parents must be a carrier, probability of a child being a carrier is 1/2. If there are affected individuals in sibship or both parents are carriers, probability of a child being a carrier is 2/3. Only applies when you know there is a possibility of homozygous affected individual, but they are not affected.
|
|
|
X-linked recessive
|
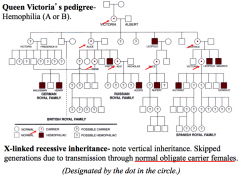
Vertical pattern of inheritance. Skipped generations (brothers, cousins, uncles, but not parents)
Females are heterozygous carriers, males are hemizygous affected. 2 mutant alleles required to cause disease in females. Common matings Heterozygous female x normal males = 1/2 affected sons, 1/2 daughters carriers Normal female x affected males = 0 sons affected, 100% daughters affected Affected males = q Affected females = q² Carrier females = 2q |
|
|
X Chromosomes
|
Share a psudoautosomal area (small area of homology that allows pairing up). Genes in this area are still active and will still result in disease
|
|
|
X-linked recessive and spontaneous new mutations
|
In a family w/no previous history of an X-linked recessive disorder, possibility that an affected individual is due to a spontaneous new mutation must be considered.
Reproductively lethal but spontaneous mutations allow it to still propagate. (Haldane principle - in pop w/ stable disease freq. rate of spontaneous new mutation = rate of loss of alleles) 1/3 of alleles lost (1 X chromo) is lost) and 1/3 of alleles new mutations. 2/3 (2 X chromor are retained) chance of carriers. In spontaneous mutations, you should not see affected individuals more than once. If you do this suggest that their mothers are carriers. |
|
|
X-linked dominant
|
Vertical pattern of inheritance. No skipped generations.
Twice as many affected females as males Common Matings Heterozygous females x normal male = 50% sons affected, 50% daughters affected Normal females x affected males = 0% sons affected, 100% daughters affected No male to male transmission. Disease in females is more severe |
|
|
Autosomal, male dominant (sex-influenced trait)
|
Both sexes have same defect, but only manifested in suceptible sex.
Balding - males are bald, females have thin hair. |
|
|
X-chromosome inactivation (lyonization)
|

Methylation is being copied as well. Daughter cells will have same inactivated X.
Pesudoautosomal reagion genes are still active and will still result in disease |
|
|
Non-Random X-inactivation
|
Structural abnormality on one X - inactivated, cells w/structural abnormality on one X are not viable → No mosacism
Balanced translocation between one X chromosome and autosome - normal X is inactivated (if translocated X is inactivated, autosome also inactivated → not viable → no mosacism |
|
|
Phenotype
|
Interaction of two alleles at a given loci
|
|
|
Dominant v. Recessive
|
Refers to interaction of alleles not traits or genes.
Dominant/Recessive disorders refers to the clinical manifestation of disease Dominant = 1 mutant allele causes disease Recessive = 2 mutant alleles cause disease |
|
|
Allelic Heterogeneity (AH)
|
Differing mutations in same alleles on same gene/loci, lead to same phenotype. Can have both dominant and recessive forms.
Causes different modes of inheritance different severity of disease CF - different mutations have different effects on protein expression/activity, but same phenotype |
|
|
AH problems
|

70% of alleles are ∆F508, 20% alleles are one of 24 common mutations, 10% of alleles are rare.
1.) Homozygous for ∆F508 = need 2 alleles = (0.7)(0.7) = 49% 2.)Heterozygous ∆F508 and other = (0.7)(0.2)(2) = 28% (For heterozygotes, must multiply by 2 since you can get from one from mother and one from father and vice versa 3.) Homozygous for other = (0.2)(0.2) = 4% 4.) Heterozygous carrier w/rare undetected alleles = (0.9)(0.1)(2) = 18% 5.) Homozygous unaffected = (0.1)(0.1) = 2% |
|
|
Dominant Negative
|
Occurs in Allelic heterogeneity
Dimerization between mutant form and normal form - leads to inactivation of normal form. Affected person having child = 50% of having affected child |
|
|
Locus Heterogeneity (LH)
|
Mutations in different alleles, different loci, cause the same disease (occurs in multi-protein complexes or when proteins/enzymes affect same pathway)
Hemophilia - mutations in diff. genes cause same phenotype - A = mutation in factor VIIIa - B = mutation in factor IXa - Both are part of VIII complex and indistinguishable clinically. Important to know which type so you give correct clotting factor |
|
|
LH problems
|

No affected daughter - carrier of 2 different mutations = no phenotype
(chance of getting affected from mother X, 1/2)(1/2 normal son)(1/2 affected son) = 1/8 |
|
|
Penetrance (P)
|
All or none phenomenon = frequency of expression of a dominant disorder in obligate heterozygote
Non-penetrant - unaffected heterozygote w/affected offspring Retinoblastoma - Mutation in tumor suppressor is not enough to cause tumor (1st hit). Must have random mutation too (2nd hit). - Homozygous for tumor suppressor mutation = death - Sporadic - need two hits in same cell - Familial - only need 1 hit, already have mutation in tumor suppressor |
|
|
P Problems
|
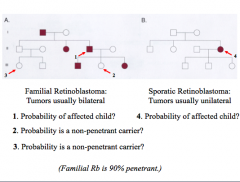
1.) (1/2)(0.9) = 45%, 0.9 = penetrance
2.) (1/2)(0.1) = 5%, 0.1 = non-penetrant 3.) (1/2)(0.1)(1/2)(0.1) = 1/400, (from dad)(non-penetrant)(chance of having allele)(non-penetrant) 4.) no greater risk = spont. mutation |
|
|
Variable Expressivity (VE)
|
Pentrance is complete, but severity is variable (mildly affected parent → severely affected child)
|
|
|
Pleiotropy
|
Diverse affects of a single gene on several organ systems and functions
|
|
|
Dominant Disorders w/no Previous Family History
|
New mutations - passed from one generation to the next
Germline mosaicism - parent is mosaic for mutation. mutation found in germline cells, but in a few of the somatic cells Delayed Age of Onset - can have children and die of an unrelated cause before manifestation of the disorder |
|
|
New mutation Problem
|

Low
1/2 3/4 affected, 2/3 affected live, 1/3 normal |
|
|
Germline Mosaicism Problem
|
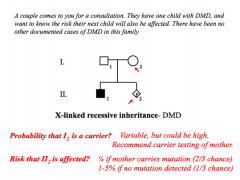
Risk for carrier parent - equal to proportion of germ cells in parent that carries the mutation
Recurrence risk for affected child - 50% chance the affected individual will pass on affected allele DMD = 1-5% recurrence risk in "de novo" due to possibility of germline mosaicism |
|
|
Delayed Age of Onset Problem
|
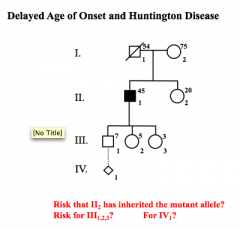
II2 = dependent on carrier status
III 50% IV1 = 1/4 |
|
|
Sequence Analysis
|
Nucleotide sequence is determined from segment of DNA, usually exons and interon-exon boundaries
Identifies only specific targeted mutations w/in a given segment of DNA Don't sequence whole gene since there's not much variance in interons Detected sequence alterations may be: - Known to be pathogenic/benign (confirmed) - Predicted to be benign/pathogenic/unknown but not reported (needs confirmation) - Promotor mutation can be detected by looking at how much RNA is produced - Northern Blot If not detected: - Pt. has sequence alteration in region of gene not covered by lab's tests - Pt. has sequence alteration that can't be detected (large deletions can't be detected since chromo w/o deletion will mask the one that has the deletion. - Pt. does not have mutation in tested gene (sequence mutation may exist in another gene) |
|
|
Mutation Scanning
|
Exons are physically tested to confirm presence of a mutation before sequencing is used to pinpoint.
Makes it faster to analyze when a disorder is caused by numerous possible mutations w/in a gene - Uses conformation sensitive gel elect. CSGE, ss conformational-polymorph SSCP, denat gradient gel elect.DGGE Reduces the amount of DNA that needs to be sequenced |
|
|
Targeted Mutation Analysis
|
Tests for presence of a specific mutation, a specific type, or a set of mutations. Not used when there are a high proportion of rare alleles
If affected pt. is not detected, you must use sequence analy. or mutation scanning for mutation ID |
|
|
Deletion/Duplication Analysis
|
Detect deletions/duplications of entire exon, multiple exons, or whole gene that are not ID by sequence analy.
Uses Q/RT-PCR, multi ligation dep. probe amplif MPLA, and SNP microarrays (CGH, CHIPs) |
|
|
QT-PCR
|
aka RTQ-PCR, use of PCR to determine amt. of DNA/RNA in a sample, used to detect heterozygous deletions/duplications
Hemizygous deletion mutations can determined standard PCR, cannot determine status of females due to other X |
|
|
DNA/RNA Analysis
|
Blotting (good for large amts):
Northern: RNA, size of intact mRNA, gene expression levels - promotor mutations Southern: genomic DNA, size/presence of DNA fragments containing gene of interest PCR (good for small amts): Distinguish small differences in size |
|
|
Protein Electrophoresis
|
Electrophoretic mobility of hemoglobin - neonatal screening
Every person w/sickle cell disease has the same point mutation β-globin: Glu6Val |
|
|
Direct DNA testing
|

PCR → Dot blot → Hybridize w/ labeled allele sp. oligonucleotides (single bp diff)
1, 2, 7, 8, 10 = SCA 4, 5, 6, 9 = carrier 3 - Sickle C (shows up as normal - 1 copy normal, 1 copy sickle C = only probing SCA = sickle C won't show) |
|
|
Trisomy 18
|
Chromosomal Abnormality:
Extra Copy of 18 Mode of Inheritance: Mostly non-disjunction (rarely translocation) Clinical Symptoms: Severe growth and developmental retardation, multi-organ failure; rocker-bottom feet, clenched fist, microcephaly Addt'l Info: 1/5000 live births <10% survive past 1st yr Recurrence low (unless one of parents have balanced translocation) Diagnosis: Karyotype |
|
|
Trisomy 13
|
Chromosomal Abnormality:
Extra Copy of 13 Mode of Inheritance: Non-disjunction (5% translocation) Clinical Symptoms: Polydactyly, rocker bottom feet, microcephaly, omphalocele, multiple organ systems effected (CNS, renal, cardiac), cutis aplasia, congenital heart disease Addt'l Info: 1/10,000 Survival: 7-10 days Insensitive to triple/quad screen Diagnosis: Karyotype, FISH |
|
|
Kleinfelter 47, XXY
|
Chromosomal Abnormality:
Extra Copy of X Mode of Inheritance: Non-disjunction Clinical Symptoms: Gynecomastia, infertility, taller stature, poor muscle tone, microorchidism (small testicles), learning behavioral issues, decreased testosterone Addt'l Info: Diagnosis: Karyotype |
|
|
Turner Syndrome 45, X
|
Chromosomal Abnormality:
Missing Copy of X Mode of Inheritance: Non-disjunction Clinical Symptoms: Infertility, small breast, short stature, brown spots (nevi), low hairline, malformation of ears, congenital heart defects, elevated LH, FSH Addt'l Info: 1/2000 female live births Recurrence = very low Diagnosis: Karyotype |
|
|
Gaucher
|
Chromosomal Abnormality:
Mutation on chromosome 1q21: defect in enzyme glucocerebrosidase (breaks down fatty acid glucosylceramide) 3 types: severity depends on mutation Mode of Inheritance: Autosomal recessive Clinical Symptoms: Glucosylceramide accumulates in white blood cells, impairs organ function, cognitive impairment, bone fractures, hepatosplenomegaly, erlenmeyer flask deformity, anemia, edema, leukoctyopenia Addt'l Info: 1/500-1/1000 Ashkenazi Jewish and Eastern European populations Diagnosis: measure beta-glucocerebrosidase activity in leukocytes Genetic testing: DNA analysis (RT-PCR) Treatment: Enzyme Replacement Therapy, or Bone Marrow Transplant |
|
|
Familial Hypercholesteroleima
|
Chromosomal Abnormality:
Mutations in LDL receptor Mode of Inheritance: Autosomal dominant Clinical Symptoms: Elevated plasma cholesterol (LDL and total) levels, accelerated arthero-scleoris, distinctive cholesterol skin deposits (xanthomas), corneal arcus Addt'l Info: 1/500 Diagnosis: Serum cholesterol levels & family history |
|
|
Duchenne/Becker Muscular Dystrophy
|
Chromosomal Abnormality:
Mutations (insertions and deletions) of the dystrophin gene (dystrophin-glycoprotein complex which links cytoskeleton of muscle fibers to the ECM) Mode of Inheritance: X-linked recessive (Almost exclusively in males, female are carriers) Allelic heterogeneity Clinical Symptoms: Absence or dysfunction of dystrophin: Progressive degeneration of skeletal muscle fibers, severe pain, impaired movement, death (respiratory/cardiac failure), elevated creatine phosphokinase in blood due to degradation of muscle cells, contraction of calves Addt'l Info: DMD: 1/3500 male births; more severe (lack of functional dystrophin) 1/3 arise from spontaneous mutations BMD: 1/20,000 male births; less severe (insufficient functional dystrophin protein) Diagnosis: Serum CPK test Genetic testing: analysis of X chromosome for deletions/insertions (Xp21) |
|
|
Hemophilia
|
Chromosomal Abnormality:
Point mutation Mode of Inheritance: X-linked recessive Locus Heterogeneity Clinical Symptoms: Type A: deficiency of clotting factor VIII (most common) Type B: deficiency of clotting factor IX Both affect the intrinsic pathway at the same stage, results in same phenotype (poor clotting) Addt'l Info: 30% of cases are spontaneous mutations Type A: 1/10,000 males Type B: 1/20,000 males Diagnosis: Gene mutation analysis to confirm which factor is affected Treatment: replacement therapy |
|
|
22q Microdeletion Syndrome (DiGeorge Velocardiofacial Syndrome)
|
Chromosomal Abnormality:
De novo mutation (microdeletion on chromosome 22q) TBX1 located in this region, required for neural crest cell migration Mode of Inheritance: Autosomal Dominant – deletion in one allele, will cause disease 7% will inherit microdeletion Clinical Symptoms: CATCH-22, Cardiac abnormality, Abnormal facies (flat philtrum), Thymic aplasia, Cleft palate, Hypocalcemia, hypoparathyroidism, 22-abnormality on chromosome 22, mental retardation Addt'l Info: 1/3800 among Hispanics 1/6000 among white, black Asians Diagnosis: FISH Recurrence risk depends on if de novo mutation or inherited. Treatment: Calcium & Vitamin D |
|
|
Neurofribromatosis
|
Chromosomal Abnormality:
Type 1: Mutation at chromosome 17q11.2 (gene deletion affecting production of neurofibromin 1, a tumor suppressor) Type 2: Mutation at chromosome 22q12 affecting production of merlin (neurofibromin 2), also a tumor suppressor Mode of Inheritance: Autosomal Dominant (gene deletion in neurofibromin) Variable expressivity Germ line cell mosaicism (rare) Clinical Symptoms: Neurofibromas (abnormal growth of neural tissue throughout body), Café au lait spots, freckles on growth and axilla (armpit), optic glioma and Lisch nodules Addt'l Info: 1/3500 50% of cases arise from spontaneous mutation Neurofibromatosis affects all neural crest cells and their derivatives Whole gene deletion → more clinically significant manifestation Diagnosis: FISH |
|
|
Marfan Syndrome
|
Chromosomal Abnormality:
Mutation (most single aa changes) in connective tissue FBN1 gene chromosome 15 (q) long arm (FBN1 codes for fibrillin-1) Also insertions, deletions, or splicing errors can occur (more severe disease) Mode of Inheritance: Autosomal Dominant High penetrance, haplo-insufficiency Variable expressivity Clinical Symptoms: Defects in ocular system (dislocated lens), skeletal system (excessive linear growth/joint laxity – abnormal curvature of the spine, sunken or protruding chest), cardiovascular system (aortic dissection) Additional Information: 2-3/10,000 individuals 25% of cases due to de novo mutations System disorder of connective tissue (high degree of clinical variability) Specific test: reveals great flexibility (Steinberg sign, Walker-Murdoch sign), arm span greater than height, pectus carinatum (pigeon test) |
|
|
Sickle Cell Anemia
|
Chromosomal Abnormality:
Point mutation, single amino acid change Glu6Val resulting in mutated β-globin gene (mutated HbS) SNP – single nucleotide polymorphism Mode of Inheritance: Autosomal Recessive Clinical Symptoms: Hemolytic anemia, sickled RBCs, hypoxemia, dehydration, multiple organ damage (autosplenectomy – immune system), marrow hyperplasia, skeletal system, hyper-bilirubinemia, renal failure, leg ulcers, dactylitis (hematopoiesis in hands and feet) Additional Information: Sickle Cell Disease 1/400 African Americans Sickle Cell Trait (heterozygous) 1/12 AA Also high incidence in Mediterranean, in regions with endemic malaria Rare, but another single amino acid Glu6Lys, change can result in mutated β-globin gene (HbC) – Sickle C |
|
|
PKU
|
Chromosomal Abnormality:
Deficiency in PAH gene (mutation on chromosome 12) causes hyperphenylalaninemia Mode of Inheritance: Autosomal Recessive Allelic heterogeneity Clinical Symptoms: Mental retardation Musty odor (due to excretion of excess phenylalanine thru sweat), rashes, elevated phenylalanine, Additional Information: 1/10,000 live births PAH (phenylalanine hydroxylase) converts phenylalanine to tyrosine Similar to FAS, to have phenylalanine in their system Children born to women with PKU at high risk for developing PKU (teratogenic exposure to phenylalanine) Dietary restrictions to limit intake of phenylalanine (restriction to proteins) |
|
|
Cystic Fibrosis
|
Chromosomal Abnormality:
Mutation in gene for cystic fibrosis transmembrane conductance regulator (CFTR), 7q31.2 Mode of Inheritance: Autosomal Recessive Clinical Symptoms: Abnormal transport of chloride/sodium across epithelium = thick, viscous secretions. Sinus infections, poor growth, coughing, shortness of breath, malabsorption of nutrients, pancreatitis, lack of normal bowel movements, clubbing of fingers/toes, infertility, amenorrhea Additional Information: Caucasian of European descent = 1/25 carrier chance 1/2000-3000 live births Diagnosis: FISH, Sweat test Treatment: Most delay the decline in organ function. Pancreatic enzyme supplements, antibiotics (IV, Inhaled, p.o.), mechanical/chemical treatments to break up secretions in lungs, Lung transplant (late-progression) |