![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
82 Cards in this Set
- Front
- Back
|
zeroth law?
|
If A in thermal equilibrium with B and B is in thermal equilibrium with C, then C is is thermal equilibrium with A |
|
|
1st law? and for infinitesimal changes?
|
∆U = q + w (for a closed system ∆n = 0) dU = δq + δw |
|
|
What does internal energy depend on?
|
V and T i.e. U(V,T) |
|
|
Define enthalpy?
|
H(p,T) = U + pV |
|
|
Link the two heat capacities?
|
Cp = (∂H/∂T)p, H = U +pV => Cp = (∂(U+pV)/∂T)p => Cp = (∂U/∂T)p + ∂/∂T(pV)p ∂/∂T(pV)p = ∂/∂T(nRT) = nR (perfect gas) =>Cp = Cv + nR |
|
|
Define entropy?
|
dS = dqrev/T |
|
|
2nd Law?
|
tot = system and surroundings if not isolated tot = system if isolated |
|
|
Clausius inequality?
|
dS ≥δq/T for an irreversible process dS>δq/T for a reversible process dS = dqrev/T |
|
|
Helmholtz free energy?
|
Using dU = dq -pdV, when dV = 0, => dU = dqV => dS ≥dU/T => dU - TdS ≤ 0 Define A = U -TS => dA = dU -d(TS) = dU -TdS - SdT (dT = 0) => dA = dU -TdS |
|
|
helmholtz spontaneous change?
|
at dT = 0, dV = 0 then dA ≤0 for a spontaneous change |
|
|
Gibbs free energy?
|
dH = dqp when dp = 0 using dS≥δq/T =>dS≥dH/T => dH -TdS≤0 Define G = H - TS =>dG = dH -d(TS) = dH - TdS -SdT dT = 0 => dG = dH -TdS |
|
|
Gibbs spontaneous change? |
then requirements for spontaneous change => dG ≤ 0 |
|
|
Spontaneity for different systems?
|
All Systems: Isolated System: ∆Ssys ≥0 ∆Ssys = 0 at equilibrium Closed or Open System: dA ≤ 0, (dT = 0, dV = 0) dA = 0 at equilibrium dG ≤ 0, (dT = 0, dp = 0) dG = 0 at equilibrium |
|
|
Third law?
|
The entropy of all perfect crystalline substances is 0 at T = 0
|
|
|
Link U and S + discuss probrem?
|
=> dU = TdS - pdV (for reversible change, but applies whether reversible or not due to all being state functions) S can not be varied in a controlled way |
|
|
derive dG(T,p) (closed system)
|
H = U +pV => dH = dU +pdV + Vdp dU = TdS - pdV => dH = TdS + Vdp G = H -TS => dG = dH - TdS - SdT => dG(T,p) = Vdp - SdT (Closed system) |
|
|
dA(T,V) for a closed system? when is it used?
|
dG(T,p) is usually used, but dA(T,V) is used in statistical thermodynamics |
|
|
dG(T,p) as partial?
|
=> dG(T,p) = (∂G/∂p)T dp + (∂G/∂T)p dT (∂G/∂p)T = V, (∂G/∂T)p = -S |
|
|
Gibbs for an open system?
|
where (∂G/∂n)t,p = µ = chemical potential |
|
|
Chemical potential for a one-component system? |
dp = 0, dT = 0 =>∫dG = ∫µdn => G = nµ => Gm = µ where Gm is the molar Gibbs free energy |
|
|
Chemical potential of a perfect gas?
|
[∂/∂p(∂G/∂n)] = [∂/∂n(∂G/∂p)] => (∂µ/∂p) = (∂V/∂n) V = nRT/p =>(∂V/∂n) = RT/p =>(∂µ/∂p) = RT/p =>∫dµ = RT∫dp/p => µ = µ⁰ + RTlnp/p⁰ |
|
|
Chemical Potential Variation with p andT (one component)?
|
one component system µ = Gm => (∂µ/∂p)T = Vm => (∂µ/∂T)p = -Sm |
|
|
Derive Clapeyron equation?
|
µ(l) = µ(v) => dµ(l) = dµ(v) =>Vm(l)dp -Sm(l)dT = Vm(v)dp - Sm(v)dT =>dp/dT = ∆Sm/∆Vm ∆G = 0 (because at equilibrium) =>∆H -T∆S = 0 =>dp/dT = ∆H/T∆V |
|
|
Derive Claussius-Clapeyron equation?
|
Assume perfect gas Vm(g)=RT/p Assume Vm(g) >> Vm(l) => ∆Vm = RT/p =>dp/dT = p∆H/RT² =>1/p dp/dT = dlnp/dT = ∆H/RT² |
|
|
How can the phase boundaries be approximated when one of the phases isa vapour?
|
Clausius-Clapeyron equation |
|
|
dG for open system containing two components?
|
=> dG(p,T,nA,nB) = Vdp - SdT + µAdnA +µBdnB => dG(p,T,ni) = Vdp - SdT + ∑µidni => ∫dG = G = ∑niµi (constant p,T, composition) (State function so equ. always true) |
|
|
Define partial molar volume? partial molar Gibbs free energy?∫dG?∫dV?
|
Vi = (∂V/∂ni)p,T,nj≠i Gi = (∂G/∂ni)p,T,nj≠i ∫dG = G = ∑niµi (constant p, T, composition) ∫dV = V = ∑niVi (constant p, T, composition) |
|
|
Gibbs-Duhem eqn?
|
=> dG = ∑nidµi + ∑µidni dG(p,T,ni) = Vdp - SdT + ∑µidni => dG = ∑µidni (constant T, p) ∴=> ∑nidµi = 0 (at constant T and p) When you change the composition at T,p = 0, the chemical potentials do not vary independently |
|
|
Gibbs-Duhem eqn for binary system?
|
dµA = -(nB/nA)dµB |
|
|
Gibbs-Duhem applied to molar volume?
|
dVA = -(nB/nA) dVB |
|
|
Chemical potential of pure liquids?meaning?
|
for a liquid in equilibrium with its vapour phase: µ*A(l) = µA(g) =>µ*A(l) = µ⁰A(g) + RTln(p*A/p⁰) - (assum perfect gas for vapour) *indicates pure liquid -we can obtain chemical potential of A in its liquid phase by measuring its vapour pressure |
|
|
Chemical potential of liquid mixture (or solvents in solutions)?
|
µ*A(l) = µ⁰A(g) + RTln(p*A/p⁰) (pure liquid) µA(l) = µ⁰A(g) + RTln(pA/p⁰) (mixture or solution) substitute for µ⁰A(g) => µA(l) = µ*A(l) + RTln(pA/p*A) |
|
|
Raoult's law?
|
pA = p*A xA where xA is the mole fraction of A in the solution The law holds increasing well as xA →1 |
|
|
Define ideal solution?
|
µA(l) = µ*A(l) + RTln(pA/p*A) pA = p*A xA => µA(l) = µ*A(l) + RTln(xA) obtain chemical potential of a solvent in an ideal solution from just how much solvent there is |
|
|
Perfect gas A is gas mixture?
|
µA(g) = µ⁰A(g) + RTln(pA/p⁰) |
|
|
Gibbs energy of mixing in an ideal solution?
|
mixed: G = nAµA + nBµB = nA(µ*A + RTlnxA) + nB(µ*B + RTlnxB) =>∆G = nARTlnxA + nBRTlnxB = nRT(xAlnxA +xBlnxB) also ∆H = 0, ∴ ∆G = -T∆S |
|
|
When is Henry's law obeyed?
|
when a solution is dilute (mole fraction <<0.1) it is found that the pressure of solute B often obey's Henry's Law. |
|
|
Ideal dilute solutions?
|
solutions that obey Henry's Law |
|
|
Henry's Law?
|
where KB does not necessarily equal p*B |
|
|
Chemical potential of an ideal dilute solution? |
-where m is molarity (moles per Kg of solvent) -m⁰ is 1 molal -µ⁰B(l) = limit (µB(l) - RTln(mB/m⁰)) as xB -> 0 µ⁰B(l) does not physically exst as it is a 1 molal solution in which the molecular interecations are the same as in an infinitely dilute solution |
|
|
Why Solute has effect on solvents Tm and Tb?
|
µA(l) = µ*A(l) + RTln(xA) (ideal solution) =>µA = µ*A + RTln(xA)= µ*A + RTln(1-xB) for small xB => ln(1-xB) ≈ -xB => µA ≈ µ*A -xBRT Solutes do not dissolve in solid solvent (some exceptions) and gas interactions are much weaker so µA(s) and µA(g) are not affected by B |
|
|
What effect solute has on solvents Tm and Tb?
|
Adding a solute lowers the melting point and raises the boiling point of the ideal solution relative to the pure liquid ∆Tb < ∆Tm |
|
|
∆Tm and ∆Tb?
|
∆Tm = xBRT*²/∆fusH ∆Tb = xBRT*²/∆vapH Where T* is the phase transition temperature for the pure solvent ∆T depends only on the mole fraction of the solute not on its identity |
|
|
osmotic pressure?
|
Pure liquid has potential µ*A(l) = µ⁰A(g) + RTln(p*A/p⁰) The solute side has potential µA(l) = µ*A(l) + RTln(xA) + ΠVm where ∆p = Π = ρgh (density x gravity x height) µ*A(l) =µA(l) at equilibrium => ΠVm = -RTln(xA) |
|
|
Van't Hoff equation for osmotic pressures?
|
small xB, ln(xA) ≈ -xB, xB = nB/(nB+nA)≈ nB/nA => ΠV = nBRT |
|
|
Ideal solubility of a pure solid? |
μ*B(s) = μ*B(sol) = μ*B(l) + RTlnxB =>lnxB = (μ*B(s) - μ*B(l))/RT =-(∆fusGm)/RT =>dlnxB = -1/R (d∆fusGm/T) (∂(∆G/T)/∂T)p = -∆H/T² =>dlnxB = 1/R (∆fusH/T² dT) => lnxB = -∆fusH/R (1/T - 1/Tm) |
|
|
eutectic point (e)?
|
The minimum melting point of the mixture -isopleth at e correspondns to the eutectic composition |
|
|
lever rule?
|
Where nα is the number of mols of phase α |
|
|
What is p assuming Raoults' law holds over entire concentration range?
|
p = xAp*A + xBp*B |
|
|
Discuss mol fraction of vapour phase in a binary liquid-vapour system?
|
-xA = mol fraction in liquid phase -yA ≠ xA because vapour will be enriched in the more volatile component usually assigned A -yA = pA/p = xAp*A/(xAp*A + (1-xA)p*B) |
|
|
T-x phase diagrams and p-x diagrams?
|
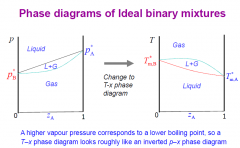
|
|
|
Gibbs phase rule?
|
F = variance (degrees of freedom) C = number of components P = numberof phases in equilibrium |
|
|
Biological standard state?
|
Labelled as µ⁰' or µ^⊕ |
|
|
∆rG⁰?
|
∆rG⁰ = -RTlnQ = -RTlnK at Equilibrium ∆rG = 0, Q = K |
|
|
∆rG?
|
= ∑niµi - ∑njµj (i = products, j = reactants) ∆rG = ∑xµi - ∑xµj (∆rG in Jmol⁻¹ )x=stoichoimetry using µA = µA⁰ + RTln(mA/m⁰) => ∆rG = ∆rG⁰ +RTlnQ |
|
|
Q?
|
= (Πprod(mj/m⁰)^vj)/(Πreact(mi/m⁰)^vi) where vx is the stoichiometry |
|
|
extent of reaction?
|
dξ = 1/vj dnj |
|
|
∆rG(ξ)? |
∆rG=dG/dξ |
|
|
Values of K?
|
Liquids: K in mole fractions, x; standard state = pure liquid Solutions: K in m/m⁰, standard state = 1 molal(with the same interactions as at infinite dilution) |
|
|
Van't hoff equation?
|
=> lnK = -∆rG⁰/RT => (∂lnK/∂T)p = -(1/R) (∂/∂T) (∆rG⁰/T)p ∂/∂T (G/T)p = -H/T² =>(∂lnK/∂T)p = ∆rH⁰/RT² |
|
|
Temperature dependence on equilibrium constant?
|
(Note for gases, K is defined at 1 bar pressure) => dlnKp/dT = ∆rH⁰/RT² |
|
|
Pressure dependence of the equilibrium constant?
|
lnK = -∆rG⁰/RT ∴ K is independent of pressure 2)For solutions we can define the standard state at any pressure (∂G/∂p)T = V => (∂lnK/∂p)T=-∆rV⁰/RT -weak dependence |
|
|
Pressure dependence of the composition at equilibrium?
|
Worked example A(g) -> B(g) + C(g), K = 0.0501, T = 900K, A initial = 1 mol increasing pressure increases A, you're going to have to look at notes for reasoning |
|
|
non-ideal systems?
|
|
|
|
Activities for different states?
|
Ideal solution; a = γx Ideal dilute solution: a = γm/m⁰ γ = activity coefficient, measures deviation from ideality, γ=1 for an ideal system (called fugacity and fugacity coefficient for gases) |
|
|
Equilibrium constant in activities?
|
K = [Πprod(ai^vi)/Πreact(aj^vj)]eq where ax = activity of x vx = stoichiometry of x NB activities still depend on the standard state, its just no longer explicit in the equation for K |
|
|
Discuss gas-liquid critical point? |
T>Tc: there is no gas-liquid phase transitions T gas phase transition will take place upon decompression inflexion appera in pV curve => dp/dVm = 0, d²p/dVm² = 0 |
|
|
Van der Waals loops?
|
|
|
|
How well does Van der Waals eqn model the system?
|
d²p/dVm² = 2RT/(Vm-b)³ - 6a/Vm⁴ = 0 => pc = a/27b², Vm,c = 3b, Tc = 8a/27Rb Zc = pcVm,c/RTc = 3/8 |
|
|
Virial equation? |
Z=pV/nRT = (1+B(n/V) + C(n/V)².....) (virial eqn) =(1+B'p+C'p²...) (∂µ/∂p) = (∂V/∂n) = ∂/∂n [(nRT/p)(1 + B'p + C'p²...)] =>µ = RT(lnp + B'p +½C'p²...) + constant p->0, µ = RTlnp + constant p->0, gases behave as perfect gases =>µ = µ⁰ +RTln(p/p⁰) =>constant = µ⁰ -RTln(p⁰) => µ = µ⁰ + RTln(p/p⁰) + RT(B'p+½C'p²....) µ = µ⁰ + RTlnf =µ⁰ + RTln(p/p⁰) + RTlnγ => lnγ = B'p +½C'p²....
|
|
|
Where does non-ideal behaviour arise from? and how is it accounted for? |
-probability of collision is proportional to the number density of the gas (n/V) -using Virial equation where: B: accounts for pairwise collisions C: acconts for 3-body collisions etc |
|
|
Boyle Temperature?
|
Z = 1 + B'p + C'p²... dZ/dp = B' + 2C'p + ... ≈ B' (at close to p=0) At Boyle Temperature (TB): B' = 0 pVm ≈ RTB (at p≈0) |
|
|
B' values?
|
Positive B': Repulsive forces dominate (high temperature) |
|
|
Fugacity?
|
-fugacity coefficients can be obtained from experimental data -as p-> 0, µreal = µideal |
|
|
activities for non-ideal solutions? |
µA(l) = µA(g) (at equilibrium) ideal: aA = xA = pA/p*A real: aA = γA(l)xA = γA(g)pA/p*A assume γA(g) = 1 => γA(l) = pA/xAp*A (γ is the ration of actual pressure to Raoult's Law pressure) |
|
|
∆mixH for regular solutions?
|
Before Hint = ½zNAεAA+½zNBεBB After Hint = ½z(NAxAεAA+NAxBεAB + ....) NA = xAN, NB = xBN, ∆mixH = ½zN(xA²εAA + xAxBεAB + xB²εBB....) write xA²= xA(1-xB), xB² = xB(1-xA) ∆mixH = ½zN(-εAA + 2εAB - εBB)xAxB ∆mixH = NβxAxB = NβxA(1-xA) β = ½z(2εAB-εAA-εBB) |
|
|
Favourable and unfavourable interactions between A and B in a regular solution?
|
β = ½z(2εAB-εAA-εBB) β<0; εAB more negative than ½(εAA + εBB) - favourable interactions between A and B - ∆mixH <0 β>0: εAB is less negative than ½(εAA + εBB) -unfavourable interactions between A and B - ∆mixH >0 |
|
|
Free energy of ∆mixG =
|
∆mixG= ∆mixH-T∆mixS ∆mixG = nβxAxB + nRT(xAlnxA +xBlnxB) |
|
|
Chemical potential of a real solution?
|
=> G = nAµA* + nBµB* +βnAnB/(nA+nB) + RT(nAln(nA/(nA+nB))+nBln(nB/(nA+nB))) =>dG/dnx =>µA = µA* + RTlnxA + βxB² =>µB = µB* + RTlnxB+ βxA² |
|
|
Temperature dependence of mixing?
|
∆mixG= ∆mixH-T∆mixS Enthalpic term is independent of temperature in the regular solution model entropic term has more weighting at higher temperatures, and so mixing is more favoured at higher T. |
|
|
spinodal?
|
line separating the unstable and meta stable regions (the line that passes through the inflection points |
|
|
binodal?
|
line separating the themodynamically stable one and two phase regions (line that passed through the minima)
|