![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
29 Cards in this Set
- Front
- Back
|
Talassemias: introdução |
Deficiência na produção de globinas. Alteração QUANTITATIVA.
A) betatalassemias B) alfatalassemias
Defeitos hereditários da hemoglobina mais comuns.
No adulto normal: - 97% hemoglobina A ou A1: ααββ - 2% hemoglobina A2: ααδδ (delta) - 1% hemoglobina F: ααγγ (gama)
- serão micro-hipo (ddx com an ferropriva) com RDW normal - há hiperferremia por mecanismos desconhecidos (assim como com an sideroblástica - fazer eletroforese de hemoglobina) *ferro sérico e ferritina altas, TIBC normal, IST alta - hemólise: aumento de LDH, redução de haptoglobina, reticulocitose - esfregaço: pontilhado basofílico, hemácias em alvo
*há proteção para Plasmodium falciparum. |
|
|
Betatalassemias: introdução |
Forma mais comum de talassemia. Principalmente gregos e italianos. Diminuição ou ausência da produção de cadeia beta. |
|
|
Betalassemias: genes |
Dois tipos de gene: - incapacidade de produzir cadeia beta (B0) - capacidade de produção limitada de cadeia beta (B+)
*gene normal seria o B
Possibilidades: - B/B: pessoa normal - B0/B0: homozigoto. Não produz nada. Talassemia major (anemia de Cooley) - B0/B+: duplo heterozigoto. Produz pouquíssimo. Talassemia major também. - B0/B e B+/B: heterozigotos. Produzem parcialmente. Assintomáticos mas com laboratório alterado. Betatalassemia minor. - B+/B+: homozigoto. Talassemia intermedia.
*gravidade definida pela quantidade de cadeia produzida. |
|
|
Betatalassemias: fisiopatologia |
(A) diminui síntese da hemoglobina: anemia com microcitose e hipocromia (B) sobram cadeias alfa, que precipitam e geram eritropoiese ineficaz (C) remanescentes da cadeia alfa geram corpúsculos de inclusão nas hemácias, tornando-as alvo da hemocaterese no baço (D) eritropoiese ineficaz aumenta absorção de ferro no intestino, gerando hemocromatose eritropoiética. |
|
|
Betatalassemias: diagnóstico |
Eletroforese de hemoglobina para todas.
Sobressairão hemoglobinas formadas por outras cadeias. HbF e HbA2.
*normal: HbA1 97%, HbA2 2%, HbF 1%. *na major: HbF chega a 90-95%, HbA1 ausente, HbA2 5-10%. *na minor: HbF de 5-20%, HbA2 de 3,5-8%. HbA1 > 70%. |
|
|
Betalassemias: major (anemia de Cooley) - clínica |
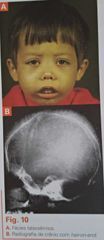
Clássico: B0/B0, mas também B0/B+.
Ausência quase completa ou completa de cadeia beta. Até 3-6m, predomina HbF sem sintomalogia (não há demanda por beta).
- A partir daí, surge anemia grave (Hb 3-5 g/dl) e icterícia. - Destruição das hemácias pelo acúmulo de cadeias alfa gera hiperplasia medular com deformidades ósseas: proeminência dos maxilares (fáscies talassêmico, fáscies de esquilo), aumento da arcada dentária e separação dos dentes e bossa frontal. - Baixa estatura é regra! Metabolismo focado em produzir hemácias. - Acompanha: litíase biliar (hemólise, cálculos pretos), hepatoesplenomegalia (pela eritropoiese extramedular e hemólise crônica). - Hemocromatose transfusional: pelo tto. Soma ao efeito da eritropoiética. |
|
|
Betalassemias: major (anemia de Cooley) - laboratório |
- Microcítica e hipocrômica. - Anemia hemolítica: reticulocitose, aumento de BI, aumento de LDH, redução da haptoglobina. - hematoscopia: hemácias em alvo (leptócitos), eritroblastose (hemácias nucleadas), pontilhado basofílico (como na anemia sideroblástica).
Conduta: não exige tratamento. Orientação e aconselhamento genético. |
|
|
Betalassemias: major (anemia de Cooley) - tratamento |
Pilar: Hipertransfusão crônica.
A depender do aumento da necessidade de hemotransfusão, realizar esplenectomia entre 6-7a. Vacinar antes para pneumococo, meningococo, hemófilo tipo B.
Hemocromatose secundária: usar quelantes como deferoxamina ou deferasirox.
"Cura": sobrevida longa livre de doença. Transplante alogênico de medula. |
|
|
Betalassemias: intermedia |
Diagnóstico frequentemente feito na adolescência ou vida adulta.
Cronicamente anêmicos: 6-9 g/dl, deformidades ósseas, litíase biliar, hepatoesplenomegalia moderada. Pode surgir hemocromatose eritropoiética. Preserva crescimento, desenvolvimento e fertilidade.
Laboratório igual à major, apenas menor grau de anemia.
Tto: - repor ác fólico - hemotransfusões para sintomáticos - quelante de ferro se ferritina muito alta |
|
|
Betalassemias: minor |
Entidade benigna.
*IMPORTANTE: grande ddx com an ferropriva, ambas hipo-micro. Aqui, RDW normal!! *cinética do ferro normal!
Traço talassêmico. Tem um gene normal e outro B0 ou B+.
Paciente assintomático, descoberta acidental. Alguns com anemia discreta (Hb > 10g/dl). Marcadores: microcitose, hipocromia, hemácias em alvo, pontilhado basofílico. |
|
|
Alfatalassemias: introdução |
Sudeste asiático e China Mecanismo genético: deleção de um ou mais genes (na betatalassemia, são mutações pontuais qualitativas). Genótipos: - aa/a_ (1 deleção): assintomático - aa/_ _ (2 deleções): alfatalassemia minor (heterozigoto a0) - a_/a_ (2 deleções): alfatalassemia minor (homozigoto a+) - a_/_ _ (3 deleções): doença da hemoglobina H - _ _/_ _ (4 deleções): hidropsia fetal Note que na alfatassemia minor, o genótipo define se há chance ou não de filhos terem hidropsia fetal... |
|
|
Alfatalassemias: hidropsia fetal |
4 depleções. Natimorto ou hidropsia fetal com morte iminente. Incompatível com vida extrauterina.
Depois de nascer, sem cadeia alfa, formam-se tetrâmeros de 4 gamas, denominada Hb Barts. Muito ávida por O2, sem liberá-lo. Gera anemia e hipóxia grave com falência cardíaca e edema fetal (hidropsia). |
|
|
Alfatalassemias: doença hemoglobina H |
3 depleções. Quadro similar à betatalassemia intermedia. Há tetrâmeros de cadeia beta (HbH) que se precipitam, mas não causam toxicidade como as alfas na betatalassemia. |
|
|
Alfatalassemias: alfatalassemia minor |
Assintomáticos, apenas com microcitose e hipocromia. Principal preocupação é orientação genética. |
|
|
Alfatalassemias: diagnóstico |
Eletroforese de hemoglobina.
Alfatalassemia minor: em geral, requer estudo genético. Eletroforese normal. Doença da hemoglobina H: Níveis de HbF e HbA2 normais e 5-40% de HbH.
Hidropsia fetal: possível diagnosticar na décima semana de gestação, por biópsia do vilo coriônico. Ausência de HbA e 80% Hb Bart. |
|
|
Alfatalassemias: tratamento |
Apenas na doença da HbH. Igual a betatalassemia intermedia. Transfusão, quelantes de ferro, esplenectomia, reposição de ác fólico. |
|
|
Mielodisplasias: introdução |
Importante causa de anemia crônica na população idosa.
Desordem adquirida. Transformação mutagênica da célula-tronco, gerando maturação anômala.
Coexistência paradoxal de medula normo ou hipercelular, associada à redução da produção das diversas linhagens hematopoiéticas. Pode ser mono, bi ou pancitopenia; pode ser tudo. *setor eritroide é o mais comprometido.
A) primária: idiopática. Geralmente > 60 anos. Fatores de risco: exposição a benzeno e radiação ambiental. B) secundária: induzida por QT/RT para neoplasias malignas. Pior prognóstico. Maior chance de evoluir para leucemia mieloide aguda. |
|
|
Mielodisplasias: clínica e laboratório |

Clínica: paciente idoso ou pós-QT/RT (mesmo jovem) com anemia normo ou macrocítica, bicitopenia ou pancitopenia, uma vez descartada a anemia megaloblástica.
Causa mais comum de óbito: infecção. Segunda mais comum: evolução para leucemia mieloide aguda.
Laboratório: - anemia sem reticulocitose - normo-normo ou macrocitose - outros achados: macrovalócitos, neutrófilos hipossegmentares e hipogranulares, sd de pseudo-Perger-Huet (neutrófilos com núcleos em forma de halter) |
|
|
Mielodisplasias: diagnóstico |

Biópsia de medula óssea. - normo ou hipercelular - diseritropoiese: núcleos megaloblásticos, hemoglobinização deficiente, sideroblastos em anel (se >15%, é anemia sideroblástica); menos de 20% de blastos também (se mais, leucemia) - localização anormal dos precursores mieloides (ALIP): mielócitos em posição central na medula (normal: paratrabecular). |
|
|
Mielodisplasias: tratamento |

Para todos, única terapia nos mais graves e/ou idosos: transfusões quando necessárias e quelantes de ferro, fatores de crescimento eritropoiético (como EPO recombinante). Terapia de baixa intensidade: para idosos com doença leve ou jovens com doença leve/moderada: azacitidina (induz maturação celular). Terapia de alta intensidade: para jovens (<60a) com com dç moderada/grave mas bom performance status: transplante de células-tronco (cura). |
|
|
Anemia sideroblástica: introdução |

Grupo de desordens que têm em comum anemia + depósitos de ferro nas mitocôndrias dos eritroblastos. Formam sideroblastos em anel (distribuição perinuclear). Pode ser hereditária ou adquirida (drogas, álcool, etc; reversíveis).
Patogênese: distúrbio da síntese do heme, desde que não seja a carência do ferro. Por ex, deficiência de protoporfirina. - prejudica síntese da hemoglobina, gerando anemia e hipocromia - acumula ferro na mitocôndria
- há estímulo à absorção intestinal de ferro, gerando excesso. Hemocromatose eritropoiética (= talassemias). |
|
|
Anemia sideroblástica: quadro clínico e laboratório |
- hemácias hipo-micro - aumento do RDW (há hemácias normocíticas e até macrocíticas) - ferro sérico alto, ferritina sérica alta, TIBC normal, IST alta. - perfil hemolítico
Suspeita para dx: hipocromia com ferro sérico alto, IST elevada e ferritina elevada.
Hemácias podem reter mitocôndrias sideroblásticas, formando corpúsculos de Pappenheimer. * sideroblastos em anel no aspirado de medula; corpúsculos de Pappenheimer no sangue periférico
Confirmação do dx: aspirado de medula óssea (mielograma): >15% de eritroblastos do tipo sideroblastos em anel. |
|
|
Anemia sideroblástica: tratamento |
A) corrigir a anemia: - forma hereditária: repor B6 (piridoxina) - forma adquirida idiopática: não costuma responder a tto. - suporte transfusional
B) corrigir ou prevenir hemocromatose: - flebotomia repetida (anemia leve) - quelante de ferro (anemia moderada ou grave) |
|
|
Anemia aplásica: introdução |
Marco: pancitopenia
Definição: pancitopenia com bx de medula óssea acelular ou hipocelular (<30% do espaço medular ocupado por células hematopoiéticas, restante são adipócitos). |
|
|
Anemia aplásica: etiologia |
- Radiação ionizante - Benzeno (atenção para exposição laborativa) - Drogas: cloranfenicol - Doença do enxerto vs hospedeiro associada à hemotransfusão - Parvovírus B19: apenas crises aplásicas transitórias em falcêmicos, ou aplasia de eritrócitos pura. - Anemia de Fanconi: causa congênita mais comum de an aplásica. Ausência de polegares, baixa estaturas, manchas café de leite. - Idiopática |
|
|
Anemia aplásica: clínica e laboratório |
Astenia + hemorragia + febre. - PANCITOPENIA + NADA
*não é esperada hepatoesplenomegalia ou linfadenopatia. Sugeririam leucemias agudas, por ex (mesma tríade acima).
Laboratório: pancitopenia. Anemia pode ser normocítica ou macrocítica. Sem reticulocitose. |
|
|
Anemia aplásica: diagnóstico |
Biópsia de medula óssea. *Aspirado (mielograma) não! - medula com menos de 30% do espaço ocupado por células hematopoiéticas. Restante é tecido adiposo. - morfologia celular normal (sem descrição de qualquer alteração, epônimos, blastos). |
|
|
Anemia aplásica: tratamento |
Terapia de suporte: transfusão conforme necessidade, e tto da neutropenia febril.
Terapia definitiva: transplante de células-tronco hematopoiéticas. Acima de 40 anos não indica transplante, por risco de complicações. Nesse caso, imunossupressão com globulina antitimócito e ciclosporina. |
|
|
Aplasia eritroide isolada |
Cursa apenas com anemia, sem outras citopenias. Causa importante: Parvovírus B19. Gera quadro transitório. |