![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
222 Cards in this Set
- Front
- Back
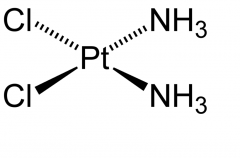
|
Cisplatin [Alkylating agent] -Admin: IV infusion -ADRs: nephrotoxic, vesicant extravasation risk (no antidote), myelosuppression (↓ wbc/rbc), otoxicity -Cytotoxic, bifunctional→ forms intrasand DNA adducts -90% CLr → high ppb -Hydrated into diaquo form -Co-admin w Amifostine (chemoprotectant) -To protect against nephrotox → admit w Cl containing solution or Sodium thiosulfate -Can use EPI, cortico, or antihists to alleviate anaphylactoid symptoms -↑ resistance →CTR1, drug trapping vesicles, conj, ↑ DNA repair
|
|
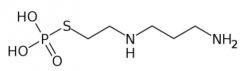
|
Amifostine [Chemoprotectant] -PRODRUG -Chloride containing solution co-admin w cisplatin to protect against toxicity -Sodium thiosulfate accumulated in renal tubule in high conc. → interacts w Cisplatin + can be excreted |
|
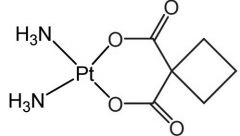
|
Carboplatin [Alkylating agent] -Cytotoxic, bifunctional → forms intrasand DNA adducts -Slow conversion to cis diaquo = 20x ↓ potent than cisplatin = ↓ ADRs -t1/2 = 3 hrs (longer than cisplatin) -Same reactive intermediate as cisplatin -ADRs: Myelosuppression -Resistance |
|
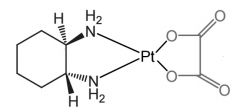
|
*Oxaliplatin [Alkylating agent] -Cytotoxic, bifunctional→ forms intrasand DNA adducts -Forms diaquo intermediate→ alkylates DNA -ADME: high Vd (440L) -↓ resistance than cisplatin/carboplatin → ↓ dependent on CTR1 active transport -Effective in patients not responding to cisplatin |
|
|
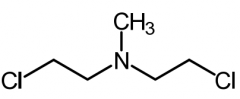
*Mechlorethamine [Nitrogen Mustard] -MOA: Cytotoxic bifunctional alkylating agent → forms intersand crosslinks -Dose: .4 mg/kg (IV ONLY) -ADRs: Potent vesicant + can cause myelogenous leukemia -Narrow margin of safety → check renal, hepatic, bone marrow |
|
|
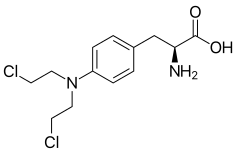
Melphalan [Nitrogen Mustard] -MOA: Cytotoxic bifunctional alkylating agent → forms intersand crosslinks -↑ stable → greater distribution into cells -ADRs: ↓ incidence, ↓ N/V vs mechlor., myelosuppression, mutagenic -Oral/IV |
|
|
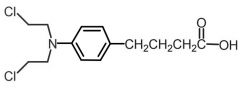
Chlorambucil [Nitrogen Mustard] -MOA: Cytotoxic bifunctional alkylating agent → forms intersand crosslinks -Oxidation → active metabolite -Good ORAL bioavailability (↓ w food) -99% ppb |
|
|
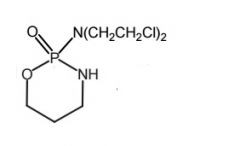
*Cyclophosphamide [Nitrogen Mustard] -MOA: Bifunctional alkylating agent → forms intersand crosslinks -PRODRUG → needs met. activation -ORAL IV -Side product: acrolein/chloroacetaldehyde can cause toxicities → Mesna (used adjunct to reduce) -USE: lymphomas, Hodgkin's disease, multiple myeloma |
|
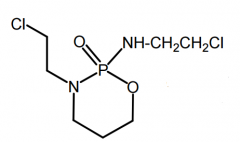
|
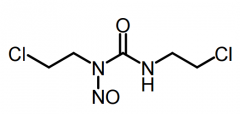
Ifosfamide [Nitrogen Mustard] -MOA: Bifunctional alkylating agent → forms intersand crosslinks -Needs met. activation → active metabolite -ADRs: Significant bladder/nephro/neuro toxicity |
|
|
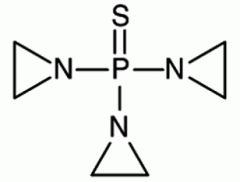
Thiotepa [Nitrogen Mustard] -MOA: Bifunctional alkylating agent → forms intersand crosslinks -Weak alkylator -Oxidative desulfuration → cytotoxic metabolite TEPA -IV only |
|
|
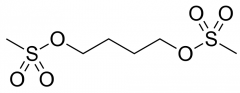
Busulfan [Nitrogen Mustard] -MOA: Monoalkylated → intra/inter adducts -ADRs: ↓ WBC count for 1 month following drug, myelosupp, BBB traversion -ORAL |
|
|
Carmustine [Nitroureas] -MOA: Produces carbocations → alkylates -USE: brain tumors, multiple myeloma (w prednisone), Hodgkin's disease, non-Hodgkin's lymphoma -ADME: Lipid soluble → traverses BBB -IV/IMPLANT -ADRs: Myelo |
|
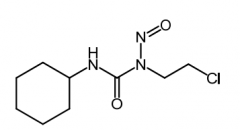
|
Lomustine [Nitroureas] -MOA: Produces carbocations → alkylates -ORAL (stable) -USE: brain tumors → highly lipid soluble + traverses BBB |
|
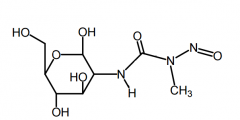
|
Streptzocin [Nitroureas] -Protonating alkylating agent -Water solube N-nitroso urea → islet specific -USE: Metastatic islet cell carcinoma -IV only -ADRs: nephrotox, diabetic neuropathy |
|
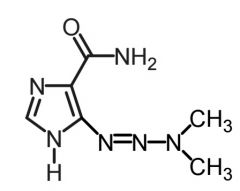
|
Dacarbazine [Nitroureas] -USE: Brain tumors -Met: CYP1A conversion→ MTIC -IV ONLY
|
|
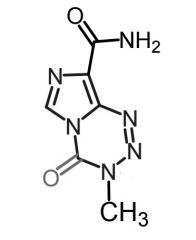
|
Temozolomide [Nitroureas] -USE: Glioblastoma multiforme/ brain tumors -Met: Hydrolytic ring opening → MTIC -ORAL/IV -ADRs: Neutropenia/cytopenia |
|
|
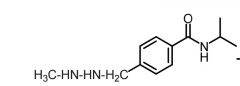
*Procarbaine [Nitroureas] -Metabolized to methyl radical → alkylates -ORAL -MOPP (Mechlorethamine + Vincristine + Prednisone + Procarbazine) → cures 70% of pts w advanced hodgkins -ADRs: Serious toxicity |
|
|
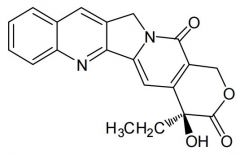
Camptothecin [Topo Poison] -MOA: Topo I inhibitor -Parent alkaloid isolated from Chinese xi shu tree -Limited water solubility→ hydroxy acid/base salt ↑ soluble but ↓ active |
|
|
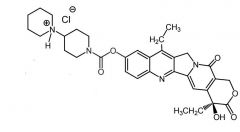
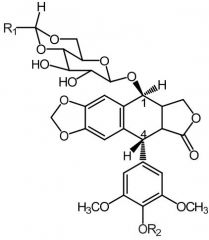
*Irinotecan [Topo Poison] -PRODRUG = active metabolite w ↑ solubility -MOA: Topo I inhibitor -ADME: glucuronidation + sulfation → SN-38 (preferential ppb) -IV -ADRs: diarrhea (dose-limiting +fatal) caused by genetic predisposition of UGT1 -USE: 1st line for MCC w 5FU + leuvoc |
|
|
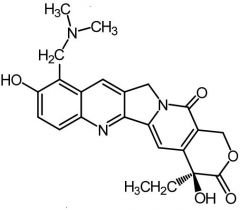
Toptecan [Topo Poison] -MOA: Topo I inhibitor -IV/ORAL -Basic side chain = ↑ solubility -ADRs: NO combo therapy w other bone marrow suppressing drugs -Renal CL |
|
|
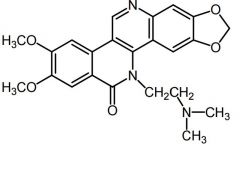
ARC-111 [Topo Poison] -MOA: Topo I inhibitor -Experimental synthetic discovered at Rutgers -Highly potent in scid mice |
|
|
Etoposide [Topo Poison] -Podophyllotoxin derivative -MOA: Topo IIa inhibitor + tubulin polym -IV/Oral -ADRs: myelosupp |
|
|
Teniposide |
[Topo Poison] -Podophyllotoxin derivative -MOA: Topo IIa inhibitor + tubulin polym -IV/Oral -ADRs: myelosupp |
|
|
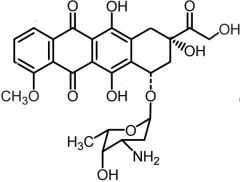
*Doxorubicin [Anthracycline] -MOA: Topo IIa inh + free radical DNA strand scission -ADRs: Cardiotoxicity due to lack of catalase by cardiac muscle -Liposoma formulation (DoxiL) → AIDs related Kaposi sarcoma + ovarian cancer (t1/2= 55 hrs) -IV |
|
|
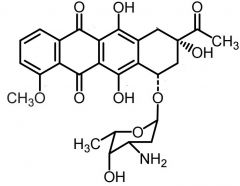
Daunorobucin [Anthracycline] -MOA: Topo IIa inh + free radical DNA strand scission -ADRs: Cardiotoxicity -USE: Nonlymphocytic cancers + AML |
|
|
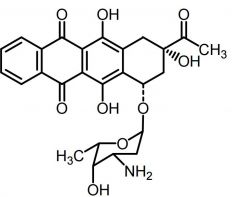
Idarubicin [Anthracycline] -MOA: Topo IIa inh + free radical DNA strand scission -ADRs: Cardiotoxicity -USE: AML |
|
|
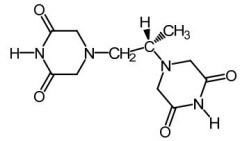
Dexrazoxane [Antioxidant]
-PRODRUG -Admin w anthracyclines -ADRs: Reduced cardiotox → hydrolyzes to amide-carboxylate + chelates iron (Fe2+) in cardiac tissue |
|
|
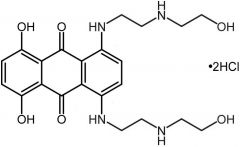
Mitoxantron [Misc. ABX] -MOA: Topo II inhibitor -ADRs: Reduced cardiotox → less prone to NADPH/CYP450 reduction -IV -USE: Acute nonlymphotoxic leukemia in combo, relapsing MS, prostate cancer |
|
|
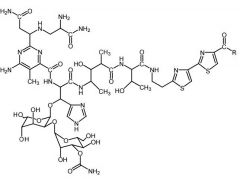
Bleomycin [Misc. ABX] -MOA: Intercalates w DNA via cytotoxic free radicals formed by chelated Fe2+ -DNA strand scission caused by OH radical -IV only -Does NOT cause bone marrow suppression |
|
|
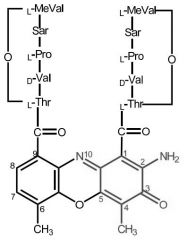
Dactinomycin [Misc. ABX] -MOA: Binds to DNA + stops transcription -Planar tricycle aromatic rings → strong binding to DNA -IV only -36 hr HL + large Vd -Toxicity + extravasation |
|
|
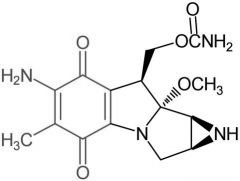
*Mitomycin [Misc. ABX] -MOA: Twofold → hydroxy radical generation + DNA alkylation -Activated by two electron bioreductive process + NQO1 reducatase → enzyme expressed in neoplastic cells |
|
|
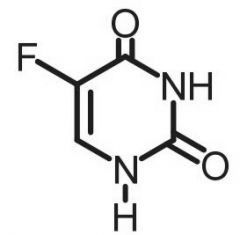
*5-fluorouracil [Antimetabolite → Pyrimidine Antag.] -MOA: Irreversible TS inhibitor → generates false substrate for TS (5-F-dUMP) + forms complex inhibiting dTMP synthesis - Also incorporates into DNA -F+ is NOT eliminated → no formation of ternary product or reaction -IV/topical -Genetic polymorphism of DPD deficiency → life threatening w ↑ exposure of drug |
|
|
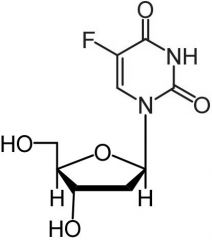
Floxuridine [Antimetabolite → Pyrimidine Antag.] -MOA: Converted in vivo to 5-F-dUMP + forms complex inhibiting dTMP synthesis -IV only -Extreme caution in renal insufficiency -PK not impacted by DPD → safer than 5-FU |
|
|
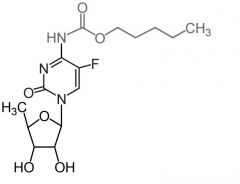
*Capecitabine [Pyrimidine Antag.]
-ORAL -PRODRUG of 5-FU → metabolized to 5-F-dUMP in vivo -ADRS: Bone marrow suppression, NVD, "hand-and-foot" syndrome -DDI: Potentially lethal w warfarin (CYP2C9 inhibition) -Oral |
|
|
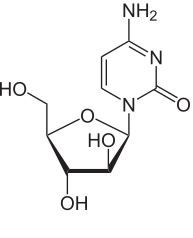
Cytarabine [Pyrimidine Antag.] -Structural difference from cytidine: Cytarabine has arabinose instead of ribose -MOA: DNA incorporation + inhibits polymerases -Active in S phase -IV only |
|
|
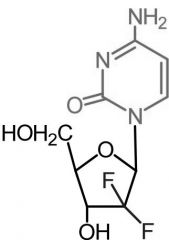
Gemcitabine [Pyrimidine Antag.] -Active form of gemcitabine → triphosphate form -MOA: DNA incorporation + DNA polym inhibitor -Longer HL > cytarabine → presence of gem-dfluoromethylene -IV only |
|
|
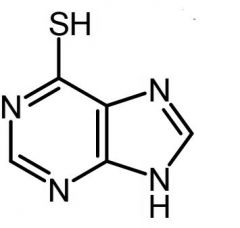
6-mercaptopurine [Antimetabolite → Purine Antag.] -MOA: Purine de novo inhibitor (AMP+GMP) -PRODRUG → converted to ribonuc by HGPRT - risk mutagenesis/malignancy vs 6-TG -Mech of resistance: 1. Uptake of nucleoside transporter 2. HGPRT enzyme activation deficiency -ADRs: TMPT deficiency→ risk for severe PURINETHOL toxicity |
|
|
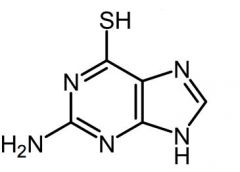
6-thioguanine [Antimetabolite → Purine Antag.]
-MOA: Purine de novo inhibitor (AMP+GMP) by inhibiting AMP transferase + DNA incorp -PRODRUG → converted to ribonuc by HGPRT -USE: Nonlymphocytic leukemias -ORAL |
|
|
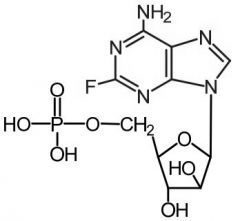
Fludarabine [Antimetabolite → Purine Antag.] -↑ aqueous solubility for IV admin → -Triphosphorylated into 2-fluoro-ara-ATP by deoxycytidine kinase -MOA: inh DNA polym + ribonuc reductase -ADRs: "AIDS in a bottle" → immunosuppressive |
|
|
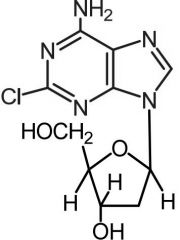
Cladribine [Antimetabolite → Purine Antag.] -MOA: Phosphorylated to 2-CdAMP→ 2-CdATP→ incorporated into DNA of dividing cells -Cell w high deoxycytidine kinase + low deoxyneucleotidase → killed by drug -IV only |
|
|
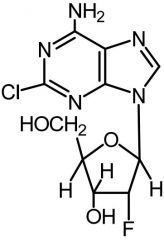
Clofarabine [Antimetabolite → Purine Antag.] -MOA: Inhibits ribonuc reductase → incorporate into DNA chain -"Tumor lysis syndrome" → disorders arising from products from dying cancer cell breakdown |
|
|
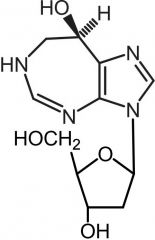
*Pentostatin [Antimetabolite → Purine Antag.] -MOA: Indirect inhibition of ribonuc reducatase by inhibiting adenosine deaminase -Advantage: In CLL → offers therapeutic efficacy comparable to fludarabine w. ↓ toxicity -No active metabolite |
|
|
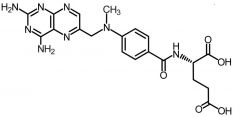
Methotrexate [Antimetabolite → Purine/Pyrim Antag.] -MOA: Twofold → inhibits DHFR + glycine amide ribonucleotide (GAR) transformylase -Polyglutamation helps toxicity of this drug against tumor cells > healthy cells -Cytotoxic against S-phase (dividing cells) -Oral/IV -Admin w leuvocorin -Resistance -USE: Breast, head neck, lung cancers + NHL + psoriasis, RA + off label MS |
|
|
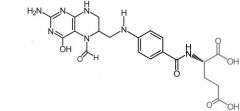
Leucovorin [Rescue Therapy] -Rescue therapy → converts DHF directly to THF -Admin w high dose methotrexate -MOA: Generates folate cofactors for continued sysnthesis of pyrimidine/purine nucleotides -ORAL/IV/IM |
|
|
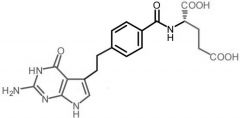
Pemetrexed [Antimetabolite → Purine/Pyrim Antag.] -MOA: inhibits DHFR and GAR transformylase → inh purine/pyrimidine synth -USE: NSCLC + malignant pleural mesothelioma (in combo w cisplatin) -IV only |
|
|
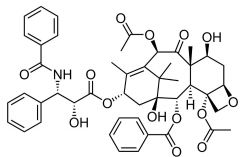
*Paclitaxel [Taxane] -MOA: Prevent depolym of microtubule → "dynamic instability" -Advantage of capectiabine combo tx: ↑ thymidine phosphorylase → ↑ capec. activity -Mech of resistance: cell efflux by Pgp substrate -IV → poor solubility -USE: 1st line→ combo tx w. cisplatin ovarian & lung cancer + breast cancer |
|
|
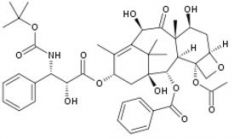
Docetaxel [Taxane] -MOA: Prevent depolymerization of microtubule → "dynamic instability" -Advantage: Better solubility bc C10-OH group → easier formulation |
|
|
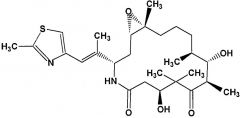
Ixabepilone [Epothilones] -Semisynthetic epo B analog MOA: Inhibits microtubule/tubulin depolym. (2x > pac.) -↑ stable than epo B → lactone replaced w lactam (not easily cleaved by esterase + ↑ stable) -USE: combo w capec. in resistant breast cancer -ADRs: peripheral neuropathy + neutropenia -BBW: for pts w impaired hepatic function -DDI: CYP3A4 |
|
|
*Vincristine [Vinca Alkaloid] -MOA: Halt cell division by inhibiting microtubule polym -ADRs: Severe vesicant -Over half the US children w cancer → give this drug |
|
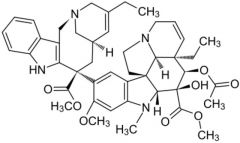
|
Vinorelbine [Vinca Alkaloid] -MOA: Halt cell division by inihibiting microtubule polymerization -ADRs: Severe vesicant -IV/oral (not available) |
|
|
*Imatinib [NRTK→ Bcr-Abl] -MOA: Type 2 NRTK (inactive enzyme) -Ph chromosome → single cause of 90% of CML -Dosing: ORAL -Develops RESISTANCE over time -ADME: F = 98%, ppb = 95%, t ½ = 18 h |
|
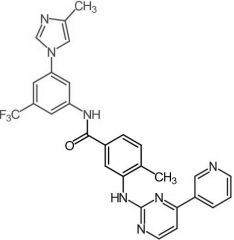
|
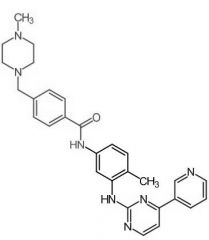
Nilotinib [NRTK→ Bcr-Abl] -MOA: Type 2 (inactive enzyme) -BBW: QT prolongation esp w CYP3A4 inh. -No Bcr-Abl mediated QT prolongation -USE: Ph+ CML in chronic phase |
|
|
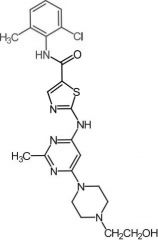
Dasatinib [NRTK→ Bcr-Abl] -MOA: Mixed type 1/2 + affinity for Src kinases -Oral (poor F) -USE: Ph+ CML, Ph+ ALL |
|
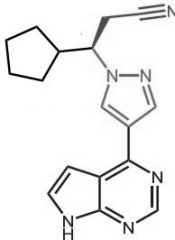
|
Ruxolitinib [NRTK→ JAK] -MOA: NRTK that transduces cytokine mediated signals by inhibiting JAK1 + JAK2 -USE: myelofibrosis + polycythemia vera -Oral |
|
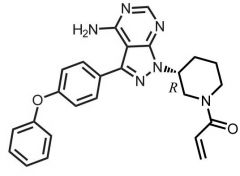
|
Ibrutinib [NTRK→ BTK] -MOA: Irreversibly inhibits BTK → ↓ survival of malignant B cell prolif + survival -Defective expression of BTK noted in ALL -USE: CLL -Oral |
|
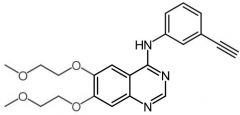
|
*Erlotinib [RTK→ EGFR1-2/HER2] -MOA: EGFR inhibitor -Oral, qd -USE: NSCLC + pancreatic cancer -Formation of electrophilic quinoneimine → fatal hepatotoxicty |
|
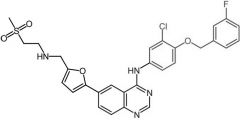
|
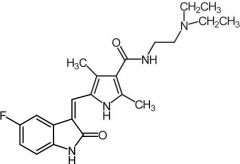
Lapatinib [RTK→ EGFR1-2/HER2] -MOA: Dual - interrupts HER2/EGFR -Oral |
|
|
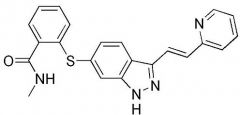
Sunitinib [RTK→ VEGFR1/2] -MOA: inhibits VEGFR (2 = more imp) → starves tumor's uncontrolled growth by retaining oxygen + nutrients -Oral, qd -Pgp substrate -USE: GI stromal tumor post imatinib failure, advanced renal carcinoma, progressive pancreatic neuroendocrine tumors |
|
|
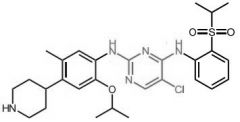
Axitinib [RTK→ VEGFR] -MOA: Inhibits VEGFR1-3 -Oral, bid |
|
|
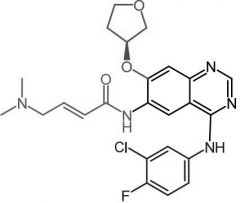
Ceritinib [RTK→ ALK] -MOA: ALK inhibitor -USE: ALK positive metastic NSCLC -Oral |
|
|
Afatinib [RTK→ MTK] -MOA: Multiple TKI inhibitor -USE: 1st line for non-small cell lung cancer |
|
|
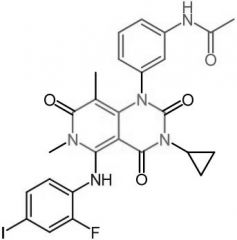
Trametinib [RTK→ MEK] -MOA: Inhibits MEK1 + MEK2 -Oral -USE: Unresectable or metastic melanoma |
|
|
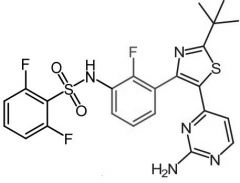
Dabrafenib [RTK→ BRAF] -MOA: Inhibitor of some mutated form of BRAF kinases -ORAL -USE: Metastic melanoma in pts w BRAF V600E/V600K mutation mono or combo w tramet -NO highly active metabolite |
|
|
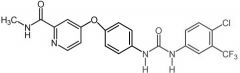
Sorafenib [RTK→ BRAF] -MOA: Inhibits CRAF, BRAF & mutant BRAF -USE: Advanced kinase cell carcinoma -Oral, bid |
|
|
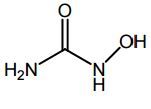
Hydroxyurea -MOA: Inhibits ribonuc reductase → inhibiting RNA to DNA conversion -Excellent ORAL bioavailability -BBW: Carcinogenic |
|
|
L-Asp & Peg-Asp
|
-L: Enzyme isolated from E.Coli -Selective killing of leukemic cells from asp plasma depletion |
|
|
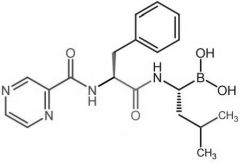
Bortezomib -MOA: Inhibits 26S proteasome → prevents targeted proteolysis→ cell death -Oral -USE: multiple myeloma |
|
|
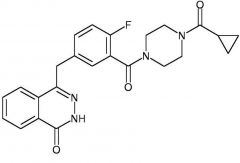
*Olaparib -MOA: Inhibits PARP enzymes (repair BRCA1/2 damaged DNA) -USE: BRCA mutated ovarian cancer -Oral |
|
|
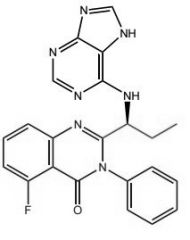
Idelalisib -MOA: Inhibits S-isoform of PI3KO, CXCR4, CXCR5 -USE: Folicular B-cell non-Hodgkin lymphoma + SLL -Oral |
|
|
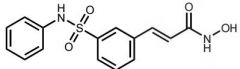
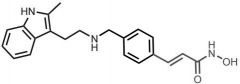
Belinostat [HDAI] -MOA: HDAI → promotes expression of p53 (tumor suppressor) -USE: peripheral T-cell lymphoma -Oral |
|
|
Panobinostat [HDAI] -MOA: HDAI -Oral -USE: multiple myleoma |
|
|
Alemtuzumab |
-Humanized -MOA: Binds to CD52 -USE: CLL + T-PLL |
|
|
Bevacizumab |
-Humanized IgG1 MAB -MOA: Inhibits VEGF-A* -USE: Metastic colon cancer in combo w standard chemo |
|
|
*Cetuximab |
-Chimeric MAB -MOA: Binds specifically to extracellular domain of EGFR -USE: K-ras* mutation (-), EGFR-expressing colorectal cancer + head, neck cancer -ADRs: Serious toxicities -First genetic test to guide treatment of cancer -KRAS mutation → poor response to cetux |
|
|
Panitumumab |
-Human MAB -MOA: Binds to EGFR* -USE: EGFR expressing metastic colorectal cancer w disease progression despite prior treatment |
|
|
Rituximab |
-Chimeric MAB -MOA: Binds to CD20 antigen → destroys B cells -USE: CLL, nonhodgekins lymphomas |
|
|
Trastuzumab |
-Humanized MAB -MOA: Binds to EGFR2 -USE: Combo w pac. for HER2 overexpressing breast cancer + gastric cancer |
|
|
Blinatumomab |
-MOA: bi-specific T-cell engager (BiTE) → links CD19 w CD3 -USE: Ph- refractory ALL |
|
|
Ramucirumab |
-Fully human MoAB -MOA: Binds to VEGFR2 → inhibiting angiogenesis USE: CLC, gastric cancer, lung cancer |
|
|
Brentuximab |
-Antibody-drug conjugate (ADC) -MOA: Attaches to CD30 → delivers MMAE (antitumor activity) -USE: ALCL |
|
|
*Ado-Trastuzumab |
-Humanized ADC → radioimmunotherapy -MOA: Binds to HER2/neu receptor -USE: HER2+ breast cancer w failed tx |
|
|
Ibritumomab |
-Murine ADC -MOA: Binds to CD20 → radiation from attached isotope kills cells + nearby cells |
|
|
Ipilimumab |
-MOA: Binds to CTLA4 → blocks CD80/86 from binding -USE: unresectable melanoma |
|
|
*Pembrolizumab |
-MOA: Blocks binding of PDL1/2 to PD1 receptor
|
|
|
Nivolumab |
-MOA: Blocks binding of PDL1/2 to PD1 receptor |
|
|
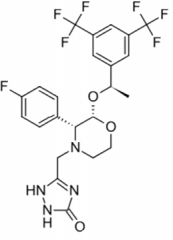
Atezolizumab |
-Humanized PDL1 MAB -MOA: Blocks PD-L1 from binding to PD-1 + B7.1 |
|
|
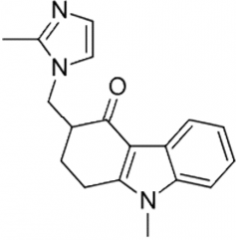
Aprepitant [Chemoprotective] -Substance P antagonist -ORAL/IV -Fosprepitant (produg) |
|
|
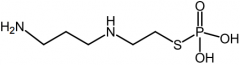
Ondansetron [Chemoprotective] -5-HT3 antagonist -ORAL/IV -ADME: F=56% |
|
|
Amifostine [Cytoprotective Agent] -↓ Cisplatin renal toxicity -IV -ADRs: hypotension |
|
|
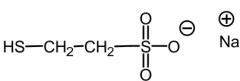
Mesna [Cytoprotective Agent] -Admin w cyclophosphadide to ↓ hemorrhagic cystitis |
|
|
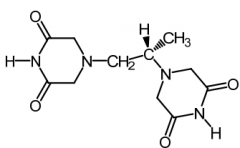
Dexrazoxane [Chemoprotective] -Admin w anthracycline (doxyrubicin) to ↓ cardiomyopathy -MOA: EDTA derivative → chelates iron + ↓ formation of superoxide radicals -IV |
|
|
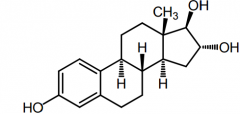
Estriol -Largest conc in urine |
|
|
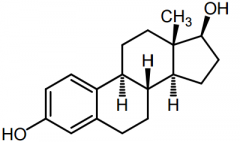
17B-Estradiol [Estrogen] -Most potent estrogen -Produced greatest in body -Conj + eliminated by intestine -Rapidly oxidized in liver |
|
|
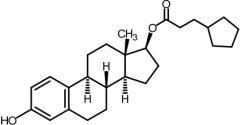
Estradiol cypionate [Estrogen] -PRODRUG of Estradiol -IM monthly -BBW→ endometrial carcinoma in post menopausal women |
|
|
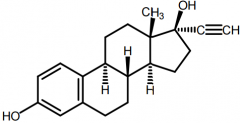
Ethynyl Estradiol [Estrogen] -Oral → rapidly + completely absorbed - > stability than estradiol -C17 prevents oxidation -First pass metabolism → enterophepatic circulation |
|
|
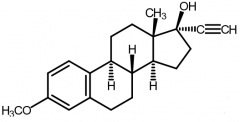
Mestranol [Estrogen]
-ORAL, inj, top -PRODRUG of EE → dealkylated in vivo + met. to EE via hepatic oxidative demethylation -Used as OC in combo w EE
|
|
|
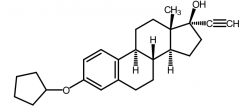
Quinestrol [Estrogen] -ORAL, inj, top -PRODRUG of EE → dealkylated in vivo + met. to EE via hepatic oxidative demethylation -Used as OC in combo w EE -Once weekly dosing |
|
|
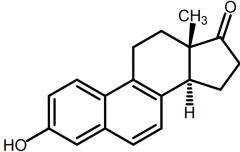
Equilenin [Conj. Estrogen] -Excreted in urine of pregant mares -In urine as Na+ sulfate conj. -Used as estro prep in combo w sodium sulfate |
|
|
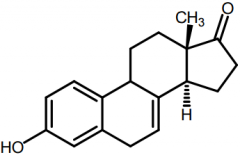
Equilin[Conj. Estrogen] -Excreted in urine of pregnant mares -In urine as Na+ sulfate conj. -Used as estro prep in combo w sodium sulfate |
|
|
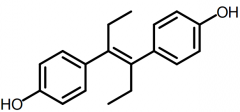
DES [Nonsteroidal Estrogen] -Active Z-isomer is only 10% active as E-isomer -Active as estrone at receptor -DC'd →birth defects + rare tumors |
|
|
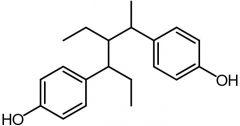
Benzestrol [Nonsteroidal Estrogen] -Same conformation of DES -Not used → same issues |
|
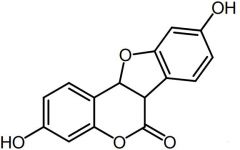
|
Coumestrol [Nonsteroidal Estrogen] |
|
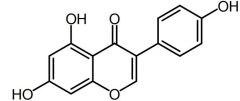
|
Genistein [Nonsteroidal Estrogen] |
|
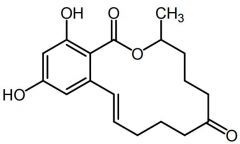
|
Zearalenone [Nonsteroidal Estrogen] |
|
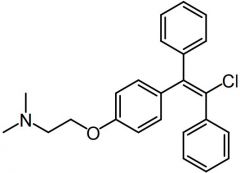
|
Enclomiphene [Estrogen] |
|
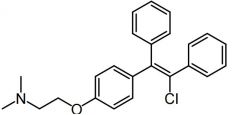
|
Zuclomiphene [Antiestrogen] |
|
|
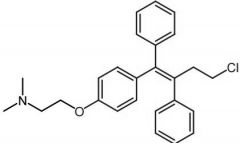
Clomiphene |
-Mixture of enclomiphene/zuclomiphene → estro/antiestro properties -Ovulation stimulatnt -Readily absorbed in GI tract -T1/2 life = 5 days |
|
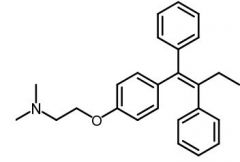
|
Tamoxifen [Antiestrogen → SERM] -Estrogen receptor antagonist in breast tissue -Metabolites: ↑ potent minor metabolite (CYP2D6) + N-des as major (CYP3A4) -QTc prolongation but no TdP -Resistance → relapse after 5 years (drug "feeds" tumor) -Oral |
|
|
Toremifene [Antiestrogen → SERM] -Mostly selective estrogen antagonist -N-desmethy toremifene → active metabolite -BBW→ QTc prolongation |
|
|
Raloxifene [Mixed SERM] -Osteoblasts/clasts → estrogen agonist -Breast/uterine → estrogen antagonist -Effective as tamox w ↓ risk |
|
|
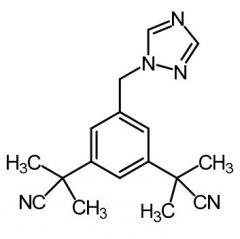
Anastrozole [Nonsteroidal Aromatase Inhibitor] -Competitive inhibitor → blocks production of estro from testosterone by aromatase (which demethylates C10 + aromatizes A ring) -ORAL -Used w out resistance limitation unlike tamox |
|
|
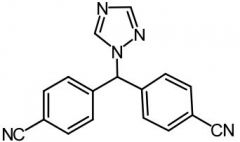
Letrozole [Nonsteroidal → Aromatase Inhibitor] -Competitive inhibitor → blocks production of estro from testosterone by aromatase (demethylates C10 + aromatizes A ring) -ORAL -Used w out limitation unlike tamox |
|
|
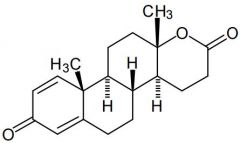
Testolactone [Aromatase Inhibitor] -Noncompetitive irreversible aromatase inhibitor -DC'd |
|
|
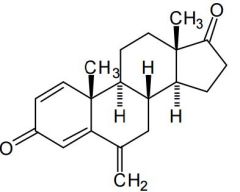
Exemestane [Aromatase Inhibitor] -Irreversible suicide inhibitor |
|
|
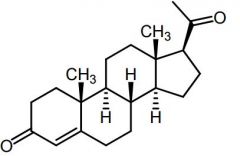
Progesterone [Progestin] -↑ efficacy + ↓ ADRs in combo w estrogen -MOA: Feedback inhibitor→ suppresses release of FSH/LH (contraceptive) -Produced in ovaries, testis, adrenal glands -Oral -Metabolites: 5B-pregnane + conj |
|
|
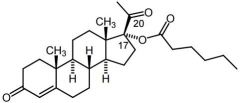
Hydroxyprogesterone [Progesterone derivative] -C17 caproate slows down C20 ketone reduction due to steric hindrance -IM -High lipophilicity → stored in fat depot + released slowly |
|
|
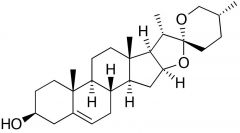
Diosgenin [Progesterone Precursor] -Converted into progesterone |
|
|
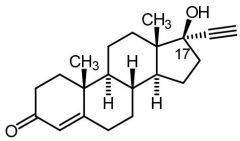
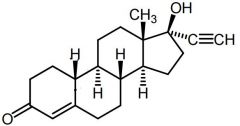
Ethisterone [1st Gen Progestin] -Plagued by androgenic activity + adrs |
|
|
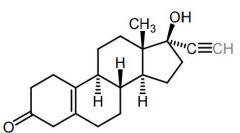
Northindrone [1st Gen Progestin] -Ethynyl group → progestational activity
|
|
|
Norethynodrel [1st Gen Progestin] -1st COCP: Norethindrone + mestranol (estrogen) held high levels of estrogen → DC'd |
|
|
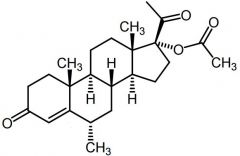
Medroxyprogesterone [2nd Gen Progestin] -MOA: Dual metabolic bloc -T1/2: 20-50 days |
|
|
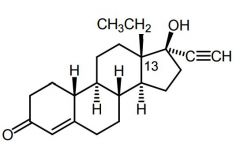
Norgestrel [2nd Gen Progestin] -↓ androgenic activity -Levonorgestrel → emergency OC |
|
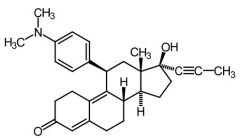
- |
Mifepristone [Progesterone Antagonist] -MOA: Binds to receptor → inhibits progesterone activity -USE: Abortifacient -Coadmin w PGE2 prostaglandins - |
|
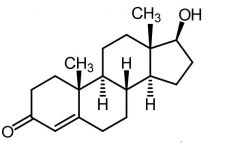
|
Testosterone [Androgen] -Two active metabolites -Involved in androgenic + anabolic activities -Binds to androgen receptors -Not used as drug → undergoes rapid metabolic oxidation -C17 group can be modified by esterification or alkylation |
|
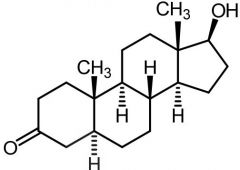
|
5a-DHT [Androgen] -10x ↑ active than testosterone -Binds to androgen receptors |
|
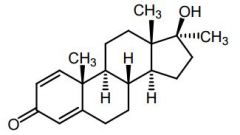
|
Methandrostenolone [Anabolic Steroid] -Has both anabolic + androgenic activity -ADRs: Nephrotox, stroke, liver tumors, jaundice, etc |
|
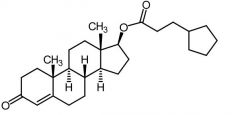
|
Testosterone Cypionate -Long acting intramuscular depot preparation |
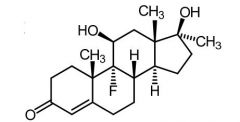
|
|
Fluoxymesterone [Anabolic Steroid] -5-10x ↑ potent than testosterone -ORAL (F=80%) - |
|
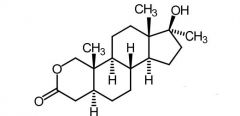
|
Oxandrolone [Anabolic Steroid] |
|
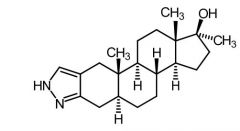
|
Stanozolol [Anabolic Steroid] |
|
|
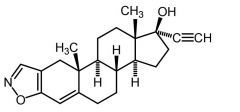
Danazol [Androgen] -Weak androgen → no estrogenic/progestin activity -USE: Endometriosis |
|
|
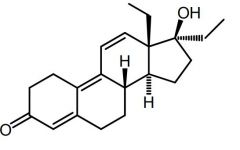
THG [Anabolic Steroid] -Designer steroid -Unstable → difficult to detect |
|
|
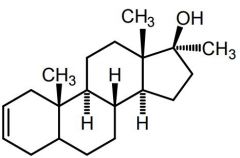
DMT [Anabolic Steroid] -Designer steroid -Unstable → difficult to detect |
|
|
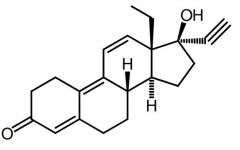
Gestrinone [Anabolic Steroid] -Designer steroid |
|
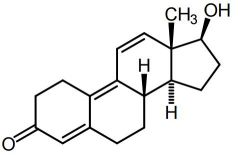
|
Trenbolone [Anabolic Steroid] -Designer steroid |
|
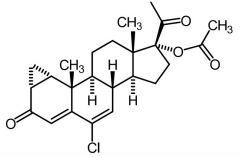
|
Cyproterone Acetate [Anti-androgen] -Competes w testosterone at androgen receptor |
|
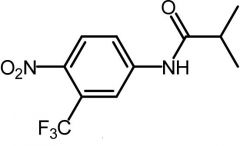
|
Flutamide [Nonsteroidal Antiandrogens] -Competes w testosterone at androgen receptor -Replaced by newer drugs |
|
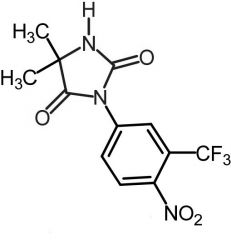
|
Nilutamide [Nonsteroidal Antiandrogen] -Competes w testostereone at androgen receptor -Replaced flutamide due to better ADR profile |
|
|
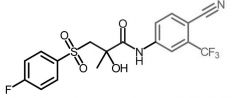
Bicalutamide [Nonsteroidal Antiandrogen] -Competes w testostereone at androgen receptor -Replaced flutamide due to better ADR profile |
|
|
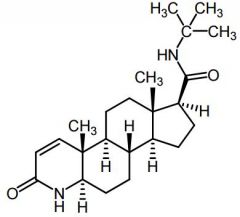
Finasteride [5a-reductase Inhibitors] -MOA: Competitive inhibitor of type 2 5a-reductase inhibitor |
|
|
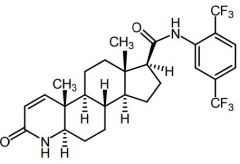
Dutasteride [5a-reductase Inhibitor] -MOA: Competitive inhibitor of type 1 + 2 5a-reductase inhibitor |
|
|
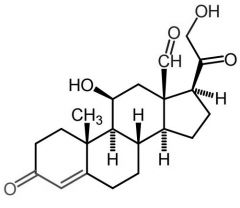
Aldosterone [Mineralocorticoid] -Naturally occuring -ADRs: loss of body Na + K, low BP, dark pigmentation |
|
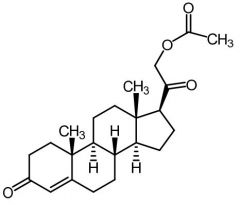
|
11-Deoxycorticosterone Acetate [Mineralocorticoid]
-Naturally occuring -no gluc activity -30x ↓ active than aldosterone -After hydrolysis - precursor to aldosterone |
|
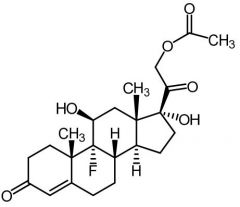
|
Fludrocortisone [Mineralocorticoid]
-Synthetic -Water soluble -ORAL -Don't need to coadminister glucs.- has some activity |
|
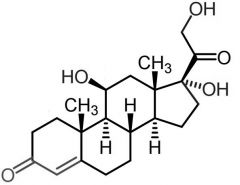
|
Hydrocortisone [Glucocorticoid] -Short acting -Affect metabolism→ utilize carbs, fats, proteins -Anti-inflammatory → inhibit transcription of + block synthesis cytokines -Major glucocorticoid found in humans -9a-Halide ↑ activity - withdraws electron density from 11-OH |
|
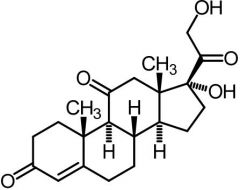
|
Cortisone [Glucocorticoid] -Short acting -Affect metabolism → utilize carbs, fats, proteins -Anti-inflammatory → inhibit transcription of + block synthesis cytokines |
|
|
Prednisone -Intermediate acting |
|
|
Prednisolone -Intermediate acting |
|
|
Methylprednisone -Intermediate acting |
|
|
Triamcinolone -Intermediate acting |
|
|
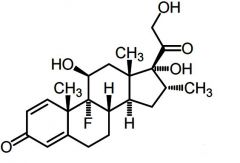
Dexamethasone -Long acting |
|
|
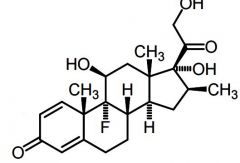
Betamethasone -Long acting |
|
|
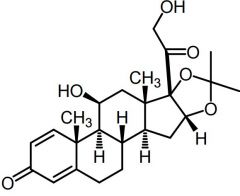
Desonide -Low mineral activity -↑ gluc activity than hydrocortisone |
|
|
Leuprolide [GnRH Agonist]
-Nonpeptide synthetic analog of LHRH -MOA: Stimulate pituary GnRH receptors = ↑ testo + down reg of LH + FSH -SUBQ in. |
|
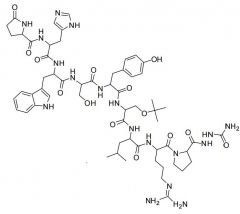
|
Goserelin [GnRH Agonist] -SUBQ inj -MOA: Binds to LHRH receptor cells = ↑ LH + sex hormones -Coadmin w bicalutamide → prevent effects of initial surge in hormone production -After 12-21 days → LH reduced enough to mimic castration |
|
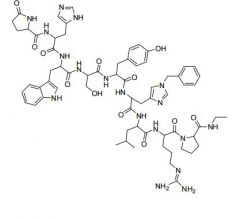
|
Histrelin [GnRH Agonist] -Nonpeptide analog of GnRH -MOA: Stimulates cells in pituitary → release LH + FSH leading to feedback inhibition causing drop in the hormones -↓ sex hormones in patients -IMPLANT |
|
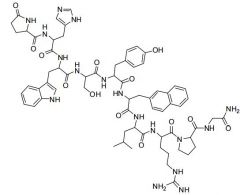
|
Nafarelin [GnRH Agonist] -Nasal spray |
|
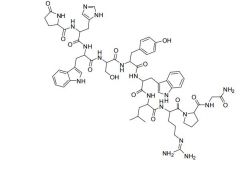
|
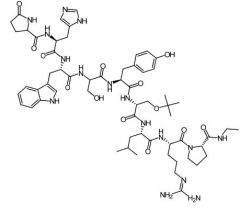
Triptorelin [GnRH Agonist] -MOA: Causes initial ↑ in testo followed by reduction to castration levels -INJECTION |
|
|
Buserelin [GnRH Agonist] -Nasal spray + parenteral formulation |
|
|
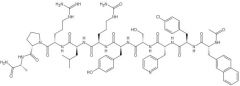
Cetrorellix [GnRH Antagonist]
-Advantage: Lacks initial flare stimulation + rapidly induce hypogonadal state |
|
|
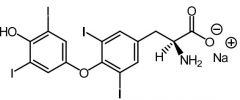
Levothyroxine [Thyroid Hormone] -Thyroxine-T4 (regulated by TSH) → converted to T3 intracellulary -Compared to T3 → longer t1/2 + ↓ potent -Major circulating form in plasma -USE: hypothyro, hashimoto, goiter -ADRs: Bone resorption + ↓ bone density w long term use |
|
|
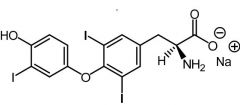
Liothyronine [Thyroid Hormone] -Triiodothyronine-T3 (regulated by TSH) -3 to 4x ↑ potent than T4 -Less used due to shorter t1/2 → multiple doses + ↑ costly -Minor circulating form in plasma -USE: W needed rapid onset or cessation of activity |
|
|
Liotrix |
[Thyroid Hormone] -Mixture of T4 + T3 (4:1) -MOA: Supplies the thyroid hormones in the ratio |
|
|
Thyroidglobin |
[Thyroid Hormone]
-From hog glands (2.5:10 ratio of T4 +T3) -Desiccated thyroid -Disadvantages → antigenicity, stability, variable hormone conc, cost |
|
|
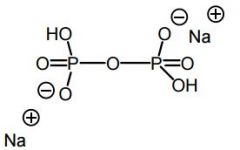
Pyrophosphate |
|
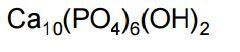
|
Hydroxyapatite |
|
|
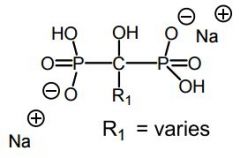
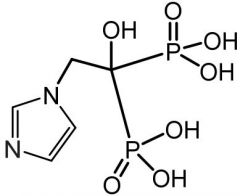
Bisphosphonates -Pyro's POP oxygen is replaced w a carbon atom → nonhydrolyzable backbone -Bind to hydroxyapatite -Inhibits osteoclast proliferation -Two types of bisphos work in different ways |
|
|
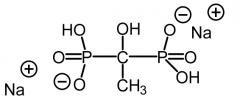
Etidronate [Non-nitrogen Bisphosphonate] -Competes with ATP + causes osteoclasts to undergo apoptosis -Leads to overall ↓ of bone breakdown |
|
|
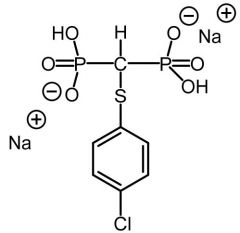
Tiludrondronate[Non-nitrogen Bisphosphonate] -Competes with ATP + causes osteoclasts to undergo apoptosis -Leads to overall ↓ of bone breakdown |
|
|
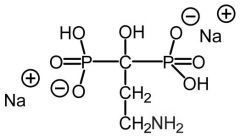
Pamidronate[Nitrogenous Bisphosphonate] -Blocks enzyme FDSP in the HMG COA pathway -HMG COA inhibition → inhibits protein prenylation in osteoclasts |
|
|
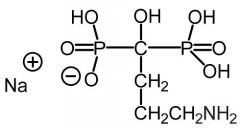
Alendronate [Nitrogenous Bisphosphonate] -Blocks enzyme FDSP in the HMG COA pathway -HMG COA inhibition → inhibits protein prenylation in osteoclasts |
|
|
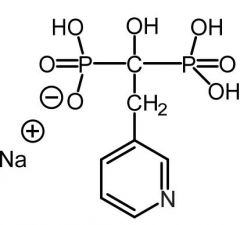
Risedronate [Nitrogenous Bisphosphonate] -Blocks enzyme FDSP in the HMG COA pathway -HMG COA inhibition → inhibits protein prenylation in osteoclasts |
|
|
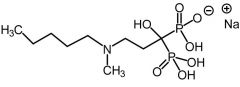
Zoledronic [Nitrogenous Bisphosphonate] -Blocks enzyme FDSP in the HMG COA pathway -HMG COA inhibition → inhibits protein prenylation in osteoclasts |
|
|
Ibandronate [Nitrogenous Bisphosphonate] -Blocks enzyme FDSP in the HMG COA pathway -HMG COA inhibition → inhibits protein prenylation in osteoclasts |
|
|
Teriparatide |
[Bone-forming Agent] -↑ # of osteoblasts -Recombinant human parathyroid hormone 1-34 -SUBQ |
|
|
Calcium salts |
[Bone-forming Agent] -↑ peak BMD + ↓ osteo risk |
|
|
Sodium fluoride |
[Bone-forming Agent] -Nonhormonal -Promotes proliferation + activity of osteoblasts -Must be couples w oral calcium supplementation |
|
|
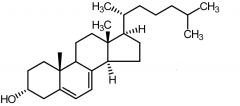
7-dehydrocholesterol [Secosteroid] -↑ intestinal absorption of Ca+ -Converted by UV radiation in skin cholecalciferol |
|
|
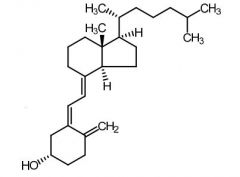
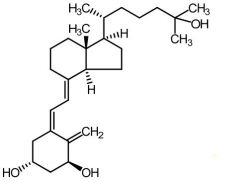
Cholecalciferol [Secosteroid] -Vitamin D3 -Transformed by Vitamin D 25 hydroxylase from liver + kidney → forms calcitrol -↑ Ca+ absorption in gut + ↓ Ca+ excretion |
|
|
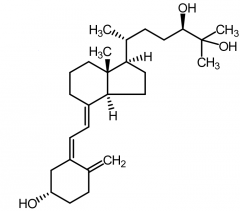
Calcitrol [Secosteroid] -Vitamin D3 metabolite -↑ bone formation |
|
|
Secalciferol [Secosteroid] -Vitamin D3 metabolite -↑ bone formation |
|
|
Lispro/Aspart/Glilisine |
[Rapid Acting Insulin] -Stay as monomers in solution -MOA: Changes made to amino acid residues in C terminue of B chain -Clear appearance -Onset: 15 min -Peak: 30-90 min -DOA: 3-4 hours |
|
|
Human Insulin
|
[Short acting] -Slow onset -Needs to be admin 30-60 min before meals -Clear appearance -Peal: 2-3 hours -DOA: 4-6 hours -Faster onset w IV admin |
|
|
Lente
|
Combined regular insulin + zinc in acetate to form a crystalloid complex that dissolves slowly in subq fluid |
|
|
Exubera |
-First inhaled insulin
-DC'd |
|
|
Afrezza |
-Rapid acting inhaled form of insulin
-Must be taken w long acting in Type 1pts -BBW: COPD/asthma pts could get bronchospasm -REMS drug |
|
|
NPH/Lente |
-Intermediate Acting Insulin
-Equal amts (+) charged polypeptid + regular insulin -Cloudy appearance -Peak: 4-12 hours -DOA: 18-24 hours -Onset: 2-4 hours |
|
|
Insulin Glargine |
-First long acting insulin
-MOA: replacement of Asn21 by glycine in A chain +addition of Arg to C terminus of B chain -SUBQ inj →microcrystals of hexamers form + dissociate into insulin monomers -Peakless profile -Onset: 1-4 hours -Peak: 5-24 hours -DOA: 20-24 hours |
|
|
Ultralente |
-Long actig insulin -Four zinc acetate crystalline product that has a even slower dissolution raate than Lente |
|
|
Insulin Detemir |
-Long acting -Results from N-acylation of the LysB29 w the 114 carbon myristic acid. -B30 Thr is absent |
|
|
Amylin |
-Co-secreted with insulin → deficient in diabetics -Inhibits glucagon secretion + delays gastic emptying + causes satiey -Unsuitable for therapeutic use→ aggregates + froms amyloid fibers-Deficiency is seen in both type 1/2 |
|
|
Pramlintide |
-Stable analog of amyline
-Structural change: Replaced the Ala25 and Serin28/29 with prolines → ↑ H2O solubility + ↓ aggregation -IM -Delays gastric emptying + suppress glucagon release + CNS anorectic effect -Coadmin w insulin for type 1 + 2 -Warning → can cause hyperglycemia in Type 1 diabetes used in combo w insulin |
|
|
Sulfonyl Ureas
|
-MOA: Stimulate insulin release + independent of plasma glucose conc→ can cause hypoglycemia
-↑ IC Ca2+ causes exocytic release of insulin -NOT EFFECTIVE in Type 1 diabetes → only effective when pt still has capacity to produce endogenous insulin |
|
|
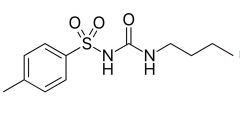
Tolbutamide [1st Gen Sulfonylurea]
-ORAL -High doses required = ↑ ADRs -Rapid metabolism (methyl converted to carboxylic acid) → short DOA + safe for elderly -Study showed ↑ mortality w long term use |
|
|
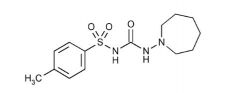
Tolazamide [1st Gen Sulfonylurea]
-Oral -Rapidly absorbed -t1/2 = 7 hrs -Can cause hypoglycemia -No accumulation upon multiple dosing -Excreted in urine |
|
|
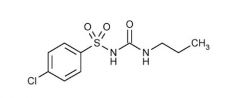
Chlorpropamide [1st Gen Sulfonylurea]
-ORAL -Cause cause hypoglycemia -Rapidly absorbed -t1/2: 36 hours→ ADR potential -ADME: oxidative metabolism + parent/metabolites excreted in urine |
|
|
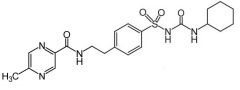
Glipizide [2nd Gen Sulfonylurea] -Better potency →50-100x ↑ active -Better therapeutically → greater binding affinity for ATP-K channel (SUR1) on B cells -ER form → ONCE daily dose -ADME: Inactive mebolites -Glucose indepent → can cause hypoglycemia -C/I: Cause hemolytic anemia + jaundice in patients w G6PD deficiency |
|
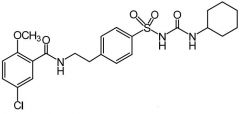
|
Glyburide [2nd Gen Sulfonylurea] -Better potency → 50-100x ↑ active -Better therapeutically → greater binding affinity for ATP-K channel (SUR1) on B cells ADRs: hypoglycemia -C/I: Glucovance (Glyburide + metformin) → BBW for lactic acidosis |
|
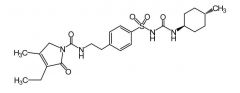
|
Glimepiride [3rd Gen Sulfonylurea] -ADME: M1 metabolite is 1/3 active as parent + M2 is inactive -Medium-long actig -Oral -ADRs→ hypoglycemia/hemolytic anemia |
|
|
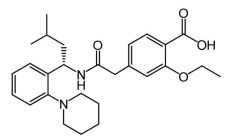
Repaglinide [Meglitinide] -MOA: binds to ATP K sensitive channels -Rapid onset + short DOA → No prolonged hyperinsulinemia, reduced weight gain + ↓ hypoglycemia -ADRS: CV→ not tissue specific, binds to SUR2A/2B on cardiac smooth muscle -Not associated w excess mortality |
|
|
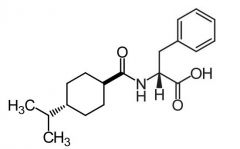
Nateglinide [Meglitinide] -MOA: binds to ATP K sensitve channels -Rapidly absorbed → rapid onset/DOA -Oral -↓ CV ADRs → much ↓ affinity for SUR1 on cardiac + skeletal muscle tissue |
|
|
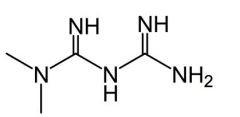
Metformin [Biguanidine] MOA: 1. Inhibits hepatic glucose synthesis 2. ↑ insulin sensitivity by ↑ uptake 3. ↓ plasma glucagon levels ↓ ADRs vs sulfonylureas → no hypoglycemia + no weight gain -Higher dose usually needed to produce clinically relevant results -Doesn't cause insulin secretion from B cells → NO hypoglycemia -Can treat insulin resistance -ADRs: Potential for rare fatal lactic acidoisis -DDIs: Cimetidine inhibits renal secretion = ↑ [metformin] in plasma |
|
|
Exenatide |
[GLP-1 Agonist]
t1/2: 3 hours (in vivo) -↑ insulin secretion + ↓ glucagon secretion -Full agonist at GLP-1 receptor + resistant to DPP-IV action -↓ HbA1c levels in sulf. treated Type 2 and appetite reduction + modest weight loss -SUBQ injection -No hypoglycemia risk w metformin combo but risk w sulfonylurea combo -ADRs: acute pancreatitis + nonfatal hemorrhagic/necrotizing panc -BBW: ER cause ↑ incidence thyroid C-cell tumors -Approved as REMS drug |
|
|
Liraglutide |
[GLP-1 Agonist] -Structure: 7-37 AA residues of GLP-1 -For weight control in pts w BMI>30 -t1/2 = 13 hours (once daily) -SUBQ inj -ADRs/BBW: thyroid tumors + pancreatitis -REMS |
|
|
Dulaglutide |
[GLP-1 Agonist] -Fusion protein -SUBQ inj pen → 1.5 mg weekly -Elim. Rate: 5 days -BBW/ADRs: Thyroid tumors/ pancreatitis -C/I in pts w Hx of thyroid/endocrine carcinoma -Approved for REMS |
|
|
Albiglutide |
[GLP-1 Agonist] -GLP-1 dimer→ resistant to DPP-IV degradation -Augments glucose-dependent insulin secretion + slows gastric emptying -SUBQ pen inj→ 30 mg weekly -Elim: 5 days -BBW: Thyroid C-cell tumors -C/I in pts w Hx of thryoid/endocrine carcinoma -Approved for REMS |
|
|
DPP-IV |
-Stimulates insulin release + inhibits glucagon release -Glucose dependent insulin secretion → less likely to cause hypoglycemia -Must have basic amino function corresponding to the penultimate form the N terminus alanine in GLP-1 |
|
|
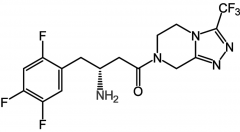
Sitagliptide [DPP-IV Inhibitor] -Glucose dependent insulin secretion → less likely to cause hypoglycemia -No effect on healthy pts→ reduces HbA1c in type 2 -2,600 fold selectivity for DPP-IV vs VIII -Shown to cause severe tox in animals -Mono or combo tx w metformin [Janumet] or sulf. for type 2 -Oral |
|
|
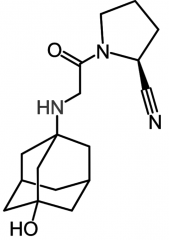
Vildagliptin [Selective DPP-IV Inhibitor]
-Glucose dependent insulin secretion → less likely to cause hypoglycemia -Dropped by Novartis -Cyano group forms covalent adduct w Ser630 → irreversibly inactivates |
|
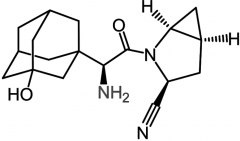
|
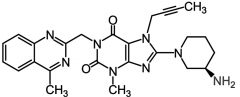
Saxagliptide [Selective DPP-IV Inhibitor] -Glucose dependent insulin secretion → less likely to cause hypoglycemia -400/75 fold selective for IV than VIII/IX -Produces glucose dependent insulin sec. -ORAL qd -Metformin combo available |
|
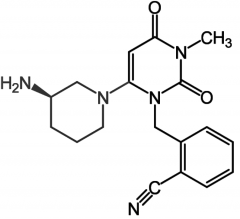
|
Linagliptide [Selective DPP-IV Inhibitor] -Glucose dependent insulin secretion → less likely to cause hypoglycemia -ORAL qd -Produces glucose dependent insulin sec. |
|
|
Alogliptide [Selective DPP-IV Inhibitor] -Glucose dependent insulin secretion → less likely to cause hypoglycemia |
|
|
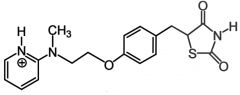
Rosiglitazone [PPAR Agonist] -Was not able to be sold w out rx → lifted in 2013 -↑ insulin sensitivity = ↑ glycemic control -Modulate gene expression → slow onset/offset -BBW: CHF, hepatotox + low density lipo. -Oral qd |
|
|
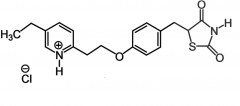
Pioglitazone [PPAR Agonist] -BBW: CHF, hepatotox + low density lipop. -Longer use (>12 mon.) associated w bladder cancer -MOA: ↑ cell responsiveness to insulin + ↓ hepatic gluc synthesis -In pts w lipid abnormalities → ↓ TGs, ↑ HDL, no change in LDL, total cholesterol -Modulate gene expression → slow onset/offset |
|
|
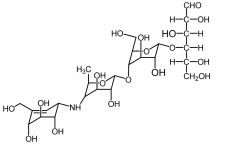
Acarbose [a-Glucosidase Inhibitor] -MOA: Competitive inhibitor → delays carb absorption in gut + postprandial hyperglycemia -Oral -Mono or combo for type 2 glycemic control -No risk of hypoglycemia/weight gain (mono) -OD will NOT result in hypoglycemia like other classes |
|
|
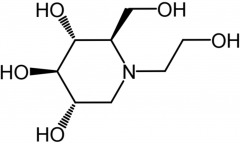
Miglitol [a-Glucosidase Inhibitor] -MOA: Delays carb absorption in gut + postprandial hyperglycemia -ORAL -Combo w sulf. → synergy for glycemic control -OD will NOT results in hypoglycemia like other classes |
|
|
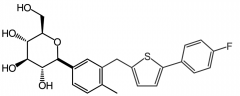
Canagliflozin [SGLT2 Inhibitor] -Glycemic control in Type 2 -Oral -ADRs: intravascular volume contraction→ hypotension |
|
|
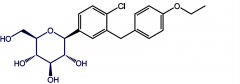
Dapagliflozin [SGLT2 Inhibitor] -ORAL 100-300 mg qd -↓ hypoglycemia + greater weight loss than sulfs -ADRs: UTI + hypotension due to intravascular volume depletion -ADME: Metabolism mediated by UGT1A9 → inactive metabolite |
|
|
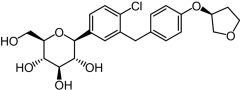
Empagliflozin [SGLT2 Inhibitor] -CV study results = ↓ rate of primary composite CVV outcome/death in type 2 -Oral, tid -ADME: Glucoronidation |
|
|
Thiaxolidinediones
|
-↑ sensitivity of cells to insulin -Interact w PPARy recepts to ↑ insulin sensititivity in adipose tissue,liver + skeletal muscle -Production of low density lipop. could be major drawback -Modulate gene expression → slow onset/offset -BBW: CHF, hepatotox |
|
|
a-glucosidase Inhibitors
|
-Inhibiting → delays process of carb absorption in gut + ↓ hyperglycemia -a gluc hydrolyzes sacchirides to glucs -OD will NOT results in hypoglycemia like other classes -Usually co admin w sulf or other secretagogues |
|
|
SGLT2 Inhibitors
|
-Glucose binds to GLUT4 → facillitates transport across membrane into cell -SGLT1 transports gluc against conc. gradient → causes reabsoprtion of gluc -Inhibitors will block the absorption of glucose |