Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
416 Cards in this Set
- Front
- Back
|
What does TPO do?
|
thrombopoietin - produces platelets
more platelets, more TPO bound, less to drive production |
|
|
Structure of platelets
|
Alpha granules: contain fibrinogen,vWF, platelet factor 4
Dense bodies: ADP - activates nieghboring platelets |
|
|
Mechanism of platelets?
|
(1)adhesion - reversible, requires vWF and collagen
(2)aggregation - irreverisble, requires fibrinogen agonist, ADP, Calcium comes in, allows binding. (3) secretion - irreversible, discharge of granules |
|
|
Adhesion of platelets?
|
subendothelium exposes vWF. vWF binds to Gp1b and connects to collagen
|
|
|
Aggregation of platelets
|
calcium binds to Gp2b/3a and increases affinity for fibrinogen. Fibrinogen cross links neighboring platelets.
|
|
|
ITP?
|
immune mediated thrombocytopenia.
IgG against own platelets. (often Gp1/2/3 receptors) Presents with petechiae or ecchymoses Acute: children get after viral rash, spontaneous recovery Chronic: need treatment Pathogenesis: anti-platelet antibodies...Fc receptor engaged in spleen, macrophages consume platelet Tx: steroids, splenectomy, immune globulin to overload Fc |
|
|
DIC
|
disseminated intravascular coagulation- bone marrow suppresion or peripheral destruction and clearance
presents with mucocutaneous hemorrhage |
|
|
IAP
|
infection associated purpura - direct vascular invasion by organism
|
|
|
TTP
|
presents with fever, anemia, thrombocytopenia, renal dysfunction, neurologic deficits
Blood: schistocytes + low platelets pathogenesis: AdamsT13 on endothelial cell. normally cleaves vWF as it is secreted. without cleaving vWF very potent aggregating. (1)congential dysfunctional AdamsT13 or (2) acquired enzyme that blocks AdamsT13 |
|
|
Neonatal period ? and hemo findings
|
0-1mo
increased retic count fetal erythrocytes have higher MCV |
|
|
Early infancy period and hemo findings
|
2-6 months
physiologic anemia: baby swithces from HbF to HbA Decreased in amount of Hb being produced, less retics |
|
|
late infancy peroid and normal hemo findings
|
6-24 months
Rapid somatic growth - need more iron adult type erythrocytes predominate |
|
|
Prematurity and heme findings?
|
physiologic anemia occurs sooner and nadir deeper. Increased risk for Fe deficiency
|
|
|
Adolescence heme
|
need more iron. gender differences appear
|
|
|
Neonatal anemias?
|
Alloimmune
Spherocytosis Elliptocytosis Pyruvate Kinase deficiency G6PD deficiency |
|
|
Alloimmune anemia of neonate
|
IgG mediated...mom antibodies to father's and babies blood type
|
|
|
Sphereocytosis of neonate
|
auto dominant.
Increased MCHC. increased osmolarity fragility |
|
|
Elliptocytosis anemia of neonate
|
auto dominant
protein spectrin defects most common |
|
|
Pyruvate kinase deficiency anemia of neonate
|
auto recessive
neonatal jaundice |
|
|
G6PD deficiency anemia of neonate
|
x linked. infection induced hemolysis. fava bean. or drug induced. have heinz body hemolytic anemia
heinz bodies on RBC |
|
|
Early infancy anemias
|
Diamond blackfan anemia
|
|
|
Diamond blackfan anemia of infancy
|
congenital pure red cell aplasia
typical facies thumb abnormalities fetal red cells, very low retics, increased HbF |
|
|
Late infancy anemias
|
(1)nutritional Fe deficit
(2) transient erythroblsatopenia of childhood (TEC) - normocytic, normochromic, low retics, very low hgb, spontaneous resolution |
|
|
Fanconi's anemia
|
pancytopenia (all increased) with congenital abnormalities
bone marrow hypoplasia radial/thumb abnorm altered skin pigment short stature |
|
|
Anemais of adolescence
|
(1)Fe deficiency
(2) secondary to disease (3) sports anemia |
|
|
Alpha thalassemias
|
(1) silent carrier - 1 deletion
(2) minor - 2 deletions (3) - H disease - 3 deletions (4) fetal hydrops - 4 deletions fatal alpha globin genes on c16 |
|
|
Beta thalassemias
|
(1)minor - microcytic, hypochromic, autosomal dominant, elevated HbA2
(2) major - transfusion dependent, autosomal recessive |
|
|
Extravascular hemolysis
|
within macrophages outside of vascular stream
RBC broken into (1) globin -> iron binds to transferrin, excess stored as ferritin (2)protoporphyrin -> bilirubin (as stones in gallbladder) -> bilirubin unconjugated -> liver -> excreted intenstive/urine |
|
|
Intravascular hemolysis
|
major red cell lysis within circulation
|
|
|
Haptoglobin function
|
created by liver. secreted. acute phase reactant. binds free hemoglobin, and in return is internalized by the hepatocyte. Low or absent Hp is indicator of intravascular hemolysis
|
|
|
excess heme clearance
|
(1)free bound to hemopexin and albumin to be taken to liver for catabolism
(2) reabsorbed in proximal tubules (can accumulate in chronic hemolysis causing renal failure) |
|
|
Direct Coombs (direct antiglobulin test{)
|
mix with antibodies against human Ig and compliment to see if antibodies are present to RBC
|
|
|
indirect coombs
|
test patients serum for antibodies...should only have antibodies to minor blood groups ABO. not Rh
ABO naturally occurring. Rh only through direct exposure |
|
|
Warm agglutinin (AIHA)
|
IgG meidated lysis of RBC
extravascular clearance through spleen |
|
|
Cold Agglutinin disease
|
IgM mediated intravascular lysis
|
|
|
Paroxysmal cold hemoglobinuria (PCH)
|
IgG mediated, acute illness often viral UTI
intravascular hemolytic anemia |
|
|
HbA1
|
alpha beta
A1c - sugar added to beta |
|
|
HbA2
|
alpha delta
|
|
|
HbF
|
alpha gamma
|
|
|
Bohr effect
|
shift right due to acidic pH
|
|
|
2,3 BPG on Po and Hgb
|
tenses Hb, shifts curve right
|
|
|
Megaloblastic anemia
|
inadequate conversion of deoxyuridylate to thymidylate.
(1) folate deficiency (2) vit b12 deficiency dyssynchrony of nucleus and cytoplasm get macrocytic RBCs and hypersegmented nuetrophils b12 converts homocyteine to methionine...folate reduces vit b12 |
|
|
Absorption of vit b12
|
intake through food, stomach frees, intrinsic factor binds, small intestine binds to intrinsic factor
|
|
|
vit b12 vs folate
|
methylmalonic acid elevated then just vit b12 deficits
homocysteine could be either |
|
|
pernicious anemia
|
autoimmune loss of effchromatin cells that produce gastric intrinsic factor
|
|
|
CML
|
chronic myelogenous leukemia:
malignant cell is myeloblast but it matures. presents: elevated WBC, but increased neutrophils, bands, metamylocytes, myelocytes, promyelos, and blasts...probably some eos and baso |
|
|
dx of chronic myelogneous leukemia
|
philadelphia chromosome: translocation 9 to 22
|
|
|
chronic myelogenous leukemia phases
|
(1) chronic phase: <5% blasts in marrow
(2) accelerated phases: 5-20% (3) blast criss >20% |
|
|
Tx of Chronic myelogenous leukemia
|
(1) high interferon
(2) allogenic stem cell (3) gleevec - tyrosine kinase inhibitor |
|
|
Chronic lymphocytic leukemia
|
chronic lymphocytic leukemia
malignant cell = more differeniated excess numbers of mature lymphocytes CBC - high lymphs present iwth anemia or thrombocytopenia immune dysregulation |
|
|
Dx Chronic lymphocytic leukemia
|
look for B cell with CD5 (t cell marker)
CD19 and Cd23 + cd5 |
|
|
Richter's transformation in Chronic lymphocytic leukemia
|
development of diffuse larger B cell lymphoma from one CLL clone
confirm with PET scan |
|
|
Hairy cell leukemia
|
very slow growing
B cell with Cd 103 (t cell marker) Dx with TRAP stain tx with cladribine |
|
|
Acute lymphocytie leukemia
|
acute lymphocytic leukemia:
epidemology: males>females present with problems with blood |
|
|
Favorable prognosis for Acute lymphocytic leukemia
|
hyperdiploidy
12->21 trans trisomy 4,10,17 |
|
|
T cell lymphoblastic leukemia/lymphoma
|
males>females
5-12 years |
|
|
B cell acute lymphoblastic leukemia/lymphoma
|
1-10 years
treat for 5 months not 2.5-3 years |
|
|
Tx of Acute lymphocytic leukemia
|
remission induction chemo: 4 weeks
intensification for 6 months continuation for 2-3 years |
|
|
Relapse Acute lymphocytic leukemia
|
testicles and CNS are sanctuaries
|
|
|
Acute myelogenous leukemia
|
acute myeloid leukemia
excess immature cells in the blood myeloblast abnormal presentation: progressive fatigue, WBC high but low ANC, rash, prolonged infection |
|
|
Dx Acute myelogenous leukemia
|
>20% blasts in the bone marrow
see auer rods |
|
|
Tx of Acute myelogenous leukemia
|
(1) induction chemo 7+3+3
cytarabine deunorubicin etoposide check BM nadir at 2 weeks (2)consolidation chemo - very high dose of cytarabine...2 doses every 12 hours on day 1, 3, and 5. Do once a month for three months |
|
|
Prognosis with Acute myelogenous leukemia
|
genetic mutations largest predictor
|
|
|
Acute promyelocytic leukemia
|
acute promyelocytic leukemia
abnormal cell: promyelocyte |
|
|
dx Acute promyelocytic leukemia nd tx
|
translocation 15 ->17
Tx: 7+4+ATRA Atra - vitamin A. induce promyelocytes to differenate |
|
|
Neutrophil development
|
myeloblast->promyeloblast-> myelocyte-> metamyelocyte ->band ->neutrophil
|
|
|
RBC development
|
proerythroblast -> basophilic eryth ->polychromatophilic erythro ->orthochromatophilic ->reticulocyte ->RBC
|
|
|
Microcytic anemias
|
decrease in hgb synthesis
Fe deficit anemia thalassemias sideroblastic anemia |
|
|
macrocytic anemia
|
defective DNA synthesis...asynchrony of nucleus and cytoplasm
Vit B12 folate (most common) inherited megaloblastic drug induced |
|
|
CINV phases
|
Acute - 1-2 hrs peaks 4-10
Delayed 1-5 hours peaks 48-72 Anticipatory phase - conditioned response |
|
|
5Ht3 antagonists
|
tx acute phase
ondansetron granisetron dolasetron palonosetron - iv only |
|
|
NK-1 antagonist
|
blocks sub P
delayed type CINV |
|
|
Dopamine Antagonists
|
phenothaizine
butyrophenones substituted benzamide |
|
|
phenothaizine
|
prochlroperazine
promethazine effective for delayed type |
|
|
butyrophenones
|
droperidol
|
|
|
substituted benzamide
|
metoclopramide
|
|
|
Benzodiazepine
|
use for anxiety associated nausea
|
|
|
corticosteroids
|
increases appetite
|
|
|
cannabinoids
|
marinol
|
|
|
general CINV guidelines
|
acute: 5Ht3 + dexamethasone + aprepitant
delayed: metoclopramide/dronabinol/ phenothiazine |
|
|
Filgrastim
|
supports proliferation of neutrophils
G-CSF use: cancer patietns with myelosuppresive chemo adverse: bone pain |
|
|
pegylated filgrastim (neulasta)
|
give as outpatient
large flat dose 24-72 hours post chemo G-CSF |
|
|
sargramostim
|
increases neutrophils, macrophages, monocytes
Use: autologous BM transplant M-CSF |
|
|
EPO
|
enhances RB production
use: non-curable pts |
|
|
Oprelvekin (neumega)
|
promotes megakaryocyte production
platelet transfusion is better |
|
|
transfusion complicatoins of platelets
|
sepsis
|
|
|
transfusion complication of RBC
|
hemolytic anemia
|
|
|
transfusion complication of plasma
|
TRALI
|
|
|
Alkylating agents mechanism of action
|
causes mistmatch in DNA
(1)template replicated misread (2)cross linking prevents unwinding (3)makes strand fragile |
|
|
Alkylating agents: names of drugs
|
(1)cyclophosphamide
(2) ifosfamide (3)platinums |
|
|
Cyclophosphamide
|
prodrug, must be converted by liver into intermediate that is then metabolized to phosphoramide mustard and acrolein
acrolein - causes hemorrhagic cystitis toxicity: myelosuppression |
|
|
Ifosfamide
|
prodrug converted to acrolein and isoforamic mustard
Toxicity: hemorrhagic cystitis |
|
|
MESNA
|
uroprotectant that binds to acrolein...must be given in advance of treatment with cyclophosphamide/ifosfamide
|
|
|
Platinum drugs and toxicities
|
cisplatin
toxicity: vomiting and nephrotoxicity carboplatin - myelosuppression oxaliplatin - cold induced neuropathy |
|
|
Amifostine
|
chemoprotectant agent for platinums
|
|
|
Anthracyclines: mechanism of action and drugs
|
(1)inhibit topo II
(2) intercalation between DNA bp blocking synthesis (3) formation of free radicals that damage DNA doxorubicin daunorubicin idarubicin epirubicin mitoxantrone Toxicity: myelosuppression doxo/daunorubicin - cardiotoxicity |
|
|
Chemo protectant for anthracyclines
|
dexrazoxane: cardioprotectant
disrupts iron antracycline complex prevents free radical formation also liposomal doxorubicin: liposomal delivery - less cardiotoxicity |
|
|
Mitoxantrone
|
second line anthracycline
side effects: blue green urine |
|
|
Antifolates:
|
methotrexate
pemetrexed |
|
|
methotrexate
|
anti folate
inhibits dihydrofolate reductase (cant make DNA bps) use: Leucovorin to rescue good cells after treatment tox: myelosuppression and mucositis nephrotoxicity (avoid NSAIDs) - prevent with alkalinizing urine |
|
|
pemetrexed
|
inhibits multiple enzymes invovled in folate metabolism
adverse: cutaneous rxns |
|
|
Pyrimidine Antagonist
|
(1)cytarabine
(2)gemcitabine (3)clofarabine (4) nelarabine (5) fluorouracil |
|
|
cytarabine
|
arabinose analog of cytosine
action: blocks DNA polymerase Tox: myelosuppression |
|
|
Gemcitabine
|
effective against solid tumors
tox: myelosuppression |
|
|
fluorouracil
|
fluorinated analog of uracil
metabolized to FdUMP which binds TS and prevents conversion of T to U Tox: myelosupprression bloody diarrhea, mucositis |
|
|
Purine Analogs:
|
action: inhibit de novo purine synthesis
(1)mercaptopurine (2) thioguanine (3)fludarabine/cladribine |
|
|
Mitotic inhibitors:
|
action: spindle poison
(1) vinca alkaloids (2) taxanes |
|
|
Vinca alkaloids
|
inhibit assembly of microtubules
(1)vincristine - neurotoxicity (2)vinblastine - myelosuppression (3) vinorelbine - myelosuppression |
|
|
Taxanes
|
make microtubules too rigid
toxicity: myelosuppression, mucositis (1)paclitaxel (2) docetaxel |
|
|
Epipodophylltoxins
|
inhibit topo II
tox: myelosuppression |
|
|
camptothecins
|
inhibit topo I
Tox: (1) topotecan - myelosuppression (2) irinotexan - diarrhea |
|
|
L-asparaginase
|
degrades asparagine
Tox: pancratitis, hypersensitivity |
|
|
Bortezomib
|
selective reversible inhibitor of proteasome
Adverse: peripheral neuropathy thrombocytopenia |
|
|
ATRA side effects
|
adverse: retinoic acid syndrome
|
|
|
Arsenic trioxide use and toxicity
|
adverse: QTC prolongation
give if retinoic acid syndrome |
|
|
Thalidomide
|
thromboembolism
|
|
|
lenalidomide
|
myelosuppression
|
|
|
HDACs action and tox
|
block HDACs to allow cell to die from normal cell regulation
(1)vorinostat - tox: dehydration, anemia, thrombocytopenia, Gi problems |
|
|
mTOR inhibitor
|
Temsirolimus - blocks translation of mRNA
Adverse: hypersensitivity, hyperglycemia, immunosuppression |
|
|
Monoclonal ABs
|
(1) Rituximab: anti CD20 for B lymphocytes
(2)Gemtuzumab ozogamicin - bone marrow suppression (3)alemtuzumab - immunosuppression |
|
|
Tyrosine Kinase Inhibitors action
|
external - ligand or receptor (Abs)
internal - binding domain (small molecules) |
|
|
Gleevec
|
tyrosine kinase inhibitor
cannot overcome T315I mutation |
|
|
Trastuzumab
|
AB to tyrosine receptor/ligand
tox: congestive heart failure |
|
|
cetuximab
|
causes acne form rash
EGFR blocker |
|
|
panitumumab
|
derm toxicity
|
|
|
erlotinib
|
acne
|
|
|
dastinib
|
nausea/vomiting
|
|
|
Fibrin Clot formation
|
thrombin converts fibrinogen to fibrin
thrombin converts 13 to 13a 13a gamma cross links, then alpha cross links fibrin |
|
|
Gamma vs alpha cross link
|
gamma is end to end (linear)
alpha 3d dimensional cross linking gamma can be digested by plasmin |
|
|
Thrombin formation:
|
TF binds to 7a
TF7a makes 9 to 9a 9a makes 10 to 10a 10a converts (2) prothrombin to thrombin |
|
|
Thrombin feedbacks to its own production:
|
thrombin converts 5 to 5a
5a makes allows 10a to convert prothrombin to thrombin faster Thrombin also makes 8 to 8a 8a is a cofactor that helps 9a convert 10 to 10a faster |
|
|
Late event in Thrombin production regulation
|
TF7a can concert 10 to 10a, bypassing 9 to 9a
Results in the amplification of fibrin clot |
|
|
Anticoagulation natural:
|
Antithrombin 3 (AT3) binds:
(1) thrombin to TAT (2) 10a to 10At (3) 9a to 9AT |
|
|
Heparin:
|
accelerates 10a to 10AT
|
|
|
Protein C system:
|
Thrombin (2a) binds to thrombomodulin on endothelial cell surface
TM2a converts protein c to aPC (protein S) aPC converts 5a to 5i (inactive) with protein S cofactor aPC converts 8a to 8i (inactive) with protein S cofactor leftover aPC binds to endothelial cell, makes tPA tPA converts plasminogen to plasmin with a fibrin cofactor plasmin converts fibrin to FDP |
|
|
Virchow's Triad of thrombosis
|
(1) alteration in the vessel
(2)alteration in the blood (3) stasis |
|
|
DIC stage 1
|
hypercoagulable state
AT3 controls coagulation cascade, but too much thrombin produced for given stimulus Increased risk of thrombosis low dose of heparin shuts down AT3 effectiveness use anti-platelet agents to block platelet aggregation |
|
|
DIC Stage 2
|
thrombin escapes because AT3 is depleted
thrombin stimulates Protein C system and down regulates 5 and 8...reduced production of thrombin tx: give platelets because they contain factor 5 and replace AT3...back to stage 1, give heparin |
|
|
DIC Stage 3
|
late/severe DIC
left over Protein C is activated by fibrinolytic system aPC inhibitor consumed, fibrinolysis activated, dissolves any fibrin clot, get free plasmin, dissolves plugs, and digests coag factors and fibrinogen Tx: give: cryo, platelets, AT3, prothrombin then give heparin to neutralize thrombin, then give AMICAR to inhibit plasma |
|
|
Impact of aging on hematopoiesis
|
bone marrow decreases in cellularity with age. however stem cells can keep up (no decrease in peripheral blood numbers).
Difference is in reserve capacity...decreases with age |
|
|
Implications of anemia in the elderly
|
incidence and prevalence increase with age.
iron deficiency, anemia of chronic diseases associated with increased mortality |
|
|
What is the relationship between age and risk of malignancy?
|
incidence of most malignancies increase with age
|
|
|
Why do elderly experience inferior outcomes from treatments?
|
1)treatment disparity - not a lot in clinical trails, also under treated (dose attenuated
2) tumor characteristics change - increased likelihood of MDR1 gene 3) host characteristics - physiologic changes, impairment in physical function, co-morbidities |
|
|
Approach to the assessment and treatment of elderly patients
|
(1)characteristics of patient - life expectancy, reserve capacity
2) characteristics of tumor 3) characteristics of treatment myelosuppresion, renal toxicity, mucositis, neurotoxicity, cardiac pretreatment |
|
|
Autologous Stem Cell Transplant
|
from the same person
cells are not cure, but rescue from the cure. if you need myelosuppressive therapy, use banked stem cells to rescue from this Requirements: chemosensitive disease healthy enough to harvest a graft Risk of mortality: 5% Negatives: more likely to relapse Indications: Multiple myeloma |
|
|
Allogenic BM transplant
|
From the same species.
Benefit: guarantee clean graft has an immunologic benefit (new cells can recognize a tumor as bad) Negatives: need to find an HLA match |
|
|
GvHD
|
T cell mediated
Risks: increased age prior exposure to blood groups CMV status Clinical: skin rash liver disorder, dairrhea |
|
|
HLA matching
|
1/4 of sibling being a match
6 genes A, B, D |
|
|
Umbilical Cord Blood transplant
|
immediate access to stem cells
less allogenic (less likely to have GvHD) less likely to be CMV positive Problem: need more cord blood for an adult cant get another collection for a new infusion |
|
|
how does radiation treatment work?
|
Damages DNA of reproducing cells
|
|
|
Types of radiation therapy
|
betatron originally
now linear particle accelerators |
|
|
Radiation use:
|
focal, local treatment for solid tumors
used often as multi-modal treatment plan |
|
|
Radiation treatment plan
|
need multiple imaging studies to determine tumor location and size.
Total dose: determined by what NORMAL cells can tolerate. give small dose over many days to weeks |
|
|
Radiation therapy
Brachytherapy |
short distance emitter for internalized treatment
|
|
|
steriotactic radiation therapy
|
use large doses for a shorter treatment interval
|
|
|
Myeloproliferative neoplasms (MPN)
|
effective and ordered hematopoiesis
cells in all stages of development |
|
|
MPN clinical features
|
cytosis of blood - elevated counts of everything
organomegaly multi-phase disease |
|
|
Chronic myelogenous leukemia
clinical |
CML - type of MPN
clinical: typically asymptomatic, large spleen, malaise, fatigue, weight loss, anemia progressive: chronic, accelerated, ends in blast (acute ML) |
|
|
Dx of Chronic myelogenous leukemia
|
philadelphia chromosome
t9.22 fusion of BCR and ABL genes ABL has tyrosine activity leukocytosis: neutrophils at all stages anemia: little room for RBC development in BM Thrombocytosis |
|
|
BM of Chronic myelogenous leukemia patients
|
hypercellular
>20% blasts |
|
|
Myelodysplastic syndrome (MDS)
|
cells do not mature effectively
|
|
|
clinical findings in Myelodysplastic syndrome
|
peripheral blood - cytopenias
no organomegaly multiphase disease that ends in BM failure older patients Anemia: mild macrocytic Thrombocytopenia Neutropenia Blasts<20% dysplastic RBC - multi-lobed nuclei Hypolobated granulocytes: pseudo pelger heut cells |
|
|
Myelodysplastic vs myeloproliferative
|
BM involved in both...hypercellular
disordered cells in dysplasia organs involved in only MPN cytopenias in MDS MPN usually translocation genetic alteration |
|
|
ways to be immuno-compromised
|
(1) ANC<500: neutropenia
(2) decreased antibody production: hypogammaglobulinemia (3)disrupted barriers (4) decreased cell mediated immunity: CD4 <200 |
|
|
Preventative measures for transmission of endogenous organisms to ICH
|
suppressive measures on host
adminster prophylaxis, clean hands |
|
|
preventative measures for transmission of exogenous organisms to ICH
|
strict hand washing most important
protective isolation special air handling systems special diets |
|
|
Initial sign of infection in ICH
|
fever, but can be due to other malignancies
|
|
|
Likely infection if host deficit is phagocyte cells
|
extra cellular pathogens
fungi, candida |
|
|
likely infection in ICH if cell mediated deficiency
|
intracellular pathogens
viruses, mycobacteria |
|
|
likely infection if ICH is antibody deficient
|
encapsulated pathogens
strep pneumo h. flu |
|
|
Transplant recipients most likely infections
|
major defect is cell mediated immunity
1st month normal immune system 2month: immuno suppressed get intracellular pathogens |
|
|
Lymph node architecture:
Cortex: |
primary and secondary follicles
b cells |
|
|
lymph node arch
paracortex |
t cells and dendritic cells
|
|
|
lymph node arch
medulla |
plasma cells and medullary sinuses
|
|
|
lymph node arch
sinuses |
macrophages
|
|
|
primary follicles in lymph node
and secondary |
naive b cells
secondary b cells introduced to antigen, begin proliferative, b cells that don't match, pushed to periphral (germinal center where active proliferation occurring) |
|
|
T cell development:
|
migrate from BM to thymus.
stop in thymic cortex. thymic epithelial cells act like Ag presenting cells. peripheral cortex has immature, rapidly proliferating cells near medulla, mature and slow proliferation 5% make it to medullla |
|
|
why do only 5% t cells make it to the medulla
|
(1) positive selection: thymic epithelial cells hsa MHC on surface and presents a peptide to T lymph. T lymph recognizes both and turns apoptosis off
(2) negative selection: central tolerance: any thymocyte that has high affinity for MHC is killed Peripheral tolerance: many tissue specific antigens are not present in thymus, encountered in peripheral, once released |
|
|
TCR determination of T cell location
|
alpha beta - stay in thymus
gamma/delta - migrate throughout body |
|
|
non-hodgkin lymphomas
|
burkitt's lymphoma
pre b cell pre t cell CLL/SLL |
|
|
Follicular Lymphoma
|
low grade nonhodgkins
increases with age morphology: small cleaved cells mature appearing CD20, CD10 Grading: determined by percentage of centrocytes to centroblasts more blasts - higher grade |
|
|
Translocation in Follicular Lymphoma
|
t14-18
|
|
|
Tx of follicular lymphoma
prognosis: |
not curable, reset clock
FLIPI scores determine prognosis adverse: >60 stage iii/IV hb<12 number of nodes >4 LDH > upper normal |
|
|
Diffuse large B cell lymphoma
|
most common intermediate grade lymphoma
Cd19/Cd10 B cell lineage, larger than normal lymphocyte abnormal architecture of lymph node no standard cytogenetics very responsive to chemo |
|
|
Tx for diffuse large b cell lymphoma
|
R CHOP
rituximab cyclophosphamide hydroxyl-doxorubicin oncovin prednisone |
|
|
Burkitt's Lymphoma
|
one of the fastest growing tumors
Types: african - jaw or face american/sporadic - GI/abdomen immunodeficient |
|
|
African burkitts lymphoma
|
malaria infection causes excess B cell production
EBV infects tumor cell Incidence 4-7 yo male>female |
|
|
american/sporadic burkitts lymphoma
|
involves abdomen, ovaries, kidneys, omentum,
only 15-30% EBV positive |
|
|
immunodeficient burkitt's lymphoma
|
HIV+ or transplants
considered defining malignancy of AIDS |
|
|
Dx of burkitt's lymphoma
|
Starry Sky pattern
cells are Cd20/CD10 |
|
|
Genetic abnormalities in burkitt's lymphoma
|
t8.14
t2.8 t8.22 |
|
|
tx of burkitt's lymphoma
|
8 months and complicated
must give CNS prophylaxis because burkitt's migrates there |
|
|
B cell differentiation...early expression of which marks?
|
TdT
|
|
|
Normal T cell immature surface marker
|
TdT
then cd4/8 |
|
|
B Cell lymphoblastic leukemia
clinical |
clinically: children and adults
predominantly leukemic agressive prognosis: cytogenetics |
|
|
b Acute lymphocytic leukemia cell phenotype
|
TdT positive
negative for cd 20 and negative sIg |
|
|
Favorable phenotype for B Acute lymphocytic leukemia
|
t12.21
hyperploidy |
|
|
unfavorable phenotype for B Acute lymphocytic leukemia
|
t9.22
t4.11 t1.19 |
|
|
T Acute lymphocytic leukemia
clinical |
adolescents
predominantly lymphomatous mediastinal mass pleural effusion |
|
|
T Acute lymphocytic leukemia phenotype
|
TdT and Cd4/CD8+
|
|
|
Chronic lymphocytic leukemia/small lymphocytic leukemia clinical
|
90% both lymphocytic and leukemia
usually asymptomatic older adults lymphocytes are small and mature Smudge cells on morphology anemia and thrombocytopenia BM shows nodular or mixed |
|
|
Small lymphocytic leukemia morphology
|
invades small lymph nodes
pseudo follicular proliferation centers SOCCER BALLL chromatin |
|
|
Small lymphocytic leukemia phenotype
|
b cell lymphoma
Cd19 Cd20 but expresses CD5 (t cell marker) |
|
|
Follicular lymphoma
clinical |
clinical: adults often asymptomatic
waxing and waning growth replace normal architecture leukemic phase - cells have prominent cleaves buckholt cells |
|
|
Follicular lymphoma phentoype
|
b cell lymphoma
cd 10, BCL2, BCL6 |
|
|
follicular lymphoma genotype
|
t14.18
BCL2 onco gene |
|
|
Diffuse large B cell lymphoma
clinical |
adults and children
rapidly enlarging mass...nodal or extranodal de novo or progression from other disease morphology: large nucleolated cells, irregular contours of nuclues |
|
|
Burkitt lymphoma
morphology phenotype |
starry sky
MIB 100% |
|
|
Burkitt lymphoma genotype
|
t8.14
t2.8 t8.22 moving of transcription factor associated with EBV |
|
|
Anaplastic large cell lymphoma
|
T cell lymphoma
children and adults nodal or extra |
|
|
Anaplastic large cell lymphoma infiltrate
|
sinusoidal infiltrate
hallmark cells |
|
|
Anaplastic large cell lymphoma phenotype
|
CD30
ALK 1protein CD3,4,43 |
|
|
Anaplastic large cell lymphoma genotype
|
TCR gene rearrangement
T2.5 |
|
|
Hodgkin lymphoma
morphology |
reed sternberg cells 1-10% of tumor cell population
inflammatory cell background |
|
|
hodgkin lymphoma phenotype
|
reed sternberg cells are CD30 and CD15 positive
EBV associated |
|
|
hodgkin lymphoma genotype
|
Ig gene rearragement
B cell lymphoma |
|
|
EPO and hypoxia
|
vHL does nto target HIF1 alpha for degradation. HIF1alpha binds HIF1beta which bind EPO. EPO-R signaling stimulates RBC production
|
|
|
Polycythemia Vera
mechanism |
clonal disorder
RBC production independent of EPO EPO low problem: mutuation in JAK2...first signallying molecule in EPO pathway. Always on |
|
|
Polycythemia Vera lab values
|
elevated hemoglobin and hematocrit
elevated RBC mass BM cellularity is increased |
|
|
Polycythemia Vera clinical findings/complaints
|
HA, visual changes dizziness, paresthesias, facial plethora
bleeding, bruising thrombosis painful red hands (erythromelaglia) |
|
|
Polycythemia Vera dx
|
increased RBC mass
platelet>400 WCC>12k low epo BM biopsy |
|
|
Polycythemia vera tx.
|
phlebotomy
hydroxyurea aspirin |
|
|
Essential thrombocythemia
mechanism |
too many platelets
clonal disoder independent of TPO often have JAK2 mutation |
|
|
Essential thrombocythemia dx
|
sustained platelet count >450
hyperplasia of megakaryocytes on bone biopsy absence of t9.22 (Test for cml) |
|
|
essential thrombocythemia
natural hx |
bleedign due to abnormal platelet function
thrombosis splenomegaly erythromelalgia |
|
|
essential thrombocythemia
tx |
hydroxyurea
aspirin |
|
|
chronic idiopathic myelofibrosis
BM results |
scarring
reticulin and or collagen fibrosis decreased cellularity dry taps |
|
|
Chronic idiopathic myelofibrosis
clinical findings |
marked splenomegaly
extramedullary hematopoiesis |
|
|
chronic idiopathic myelofibrosis
blood results |
pseudo-pelger-huet cells - neutrophils have two lobes
giant platelets anemic JAK2 mutations |
|
|
Chronic idiopathic myelofibrosis
Tx |
palliative
|
|
|
Myelodysplastic syndromes
dx |
(1)bone marrow biopsy shows too many blasts
5-19% is MDS or (2)see dysplastic cells |
|
|
Myelodysplastic syndrome prognosis
|
(1)Age
(2) IPSS scores |
|
|
Myelodysplastic 5q syndrome
|
part of 5 q missing
have more benign disease |
|
|
Plasma cell dyscarsias
types |
plasma cells grow more than they should
(1)monoclonal gammopathy of undetermined significance (MGUS) (2) multiple myeloma (3) plasma cell leukemia |
|
|
plasma cell dyscrasias dx
|
(1)measure number of place cells in BM
(2) measure amount of protein |
|
|
Lab tests - comprehensive panel for plasma cell dyscrasias
|
get albumin and protein values...protein should be too high
|
|
|
lab test for plasma cell dyscrasias quantitative immunoglobulin
|
give values of each Ig
|
|
|
lab test for plasma cell dyscrasias
SPEP |
albumin peak, alpha 1 peak, alpha 2 peak, beta peak
Gamma peak - most immunoglobulin |
|
|
lab test for plasma cell dyscrasias
IEP |
immunoglobulin electrophoresis
confirms clonality of Ig |
|
|
lab test for plasma cell dyscrasias
serum free light chains |
amount of light chains in the serum
|
|
|
Multiple myeloma - cell
|
plasma cell constantly producing Abs
nucleus off to side and flattened cytoplasm bi-phasic colors |
|
|
Multiple Myeloma clinical features
|
m>f
adults lytic bone lesions |
|
|
Multiple Myeloma lab findings
|
bence jones proteins in the urine
monoclonal serum gammopathy |
|
|
Multiple myeloma pathology
phenotype genotype |
morphology: marrow plasmacytosis >30%
cd38, cd18 genotype: Ig clonally rearranged |
|
|
Burkitt's Lymphoma genotype
|
IgH/c-myc rearrangement
t8.14 t2.8 t8.22 EBV implicated MIB 100% |
|
|
Hairy cell leukemia
clinically |
older adults
pancytopenia enlarged spleen |
|
|
Hairy cell Leukemia clinical
|
older adults
pancytopenia enlarged spleen |
|
|
hairy cell leukemia morphology
|
characteristic cytoplasmic projections
fried egg appearance dry bone tap from reticulin fibrosis express cd103,cd25,cd11c stain with TRAP |
|
|
Mycosis Fungoides morphology
|
pautrier microabscesses - lymphocytes invading dermis
lutzner cells sezary cells Tcells CD4 only (no CD7) |
|
|
Classic Hodgkin lymphoma dx
|
reed sternberg cells cd30/cd15
|
|
|
classic hodgkin lymphoma subtypes
|
(1)nodular sclerosis - fibrous bands divide into nodules, capsular fibrosis
mediastinal involvment (2)mixed cellularity - mixed inflammatory background, diffuse growth (3) lymphocyte depleted - background is small, reactive, lymphocytes (4) lymphocyte rich - sheets of RS cells, tumor necrosis |
|
|
Staging of hodgkin lymphoma
|
(1)involvement of a single node
(2) 2 or more lymph nodes on the same side of the daiphragm (3) lymph nodes on both sides of the diaphragm (4)multiple disseminated foci of involvement |
|
|
leukocytosis definition
|
absolute increase in number of leukocytes in peripheral blood
adults: greater than 10.000 |
|
|
Leukemoid reaction definition
|
excessive white blood cell response (50,000 or more)
|
|
|
Corticosteroids affect on WBC
|
decrease emigration of neutrophils from blood into tissues
increase release of mature neutrophils from the bone marrow decrease margination of neutrophils inside vasculature |
|
|
Neutrophilia definition
|
absolute neutrophil count of 8000 or higher
|
|
|
mechanisms of neutrophilia
|
(1)increased production by bone marrow
(2)increased mobilization from sotarge pool (3) failure to exit the circulation |
|
|
Leukocyte adhesion deficiency (hereditary mutation)
|
number of circulating neutrophils is increased
delayed detachment or prolonged healing of umbilical stump cant get leukocytes out of blood |
|
|
lymphocytosis definition
|
normally 20-40% of circulating WBC
(1)relative lymphocytosis results when there is a neutrophilic leukocytosis (2) absolte lymphocytosis (a) >9000 in infant (b) 7200 older children (c)>4000 in adults |
|
|
absolute vs relative lymphocytosis
|
absolute - acute infection, chronic infections
relative - normla in young, during viral infections, splenomegaly |
|
|
benign reactive lymphocytosis
|
pronounced in pertusis, acute infectious lymphocytosis, infectious mono
|
|
|
eosinophilia definition
|
usually less than 5%
storage in pools 5 times larger play role in phagocytizing antigen-antibody |
|
|
causes of eosinophilia
|
allergic, parasitic infections
|
|
|
causes of basophilia
|
viral infections, chronic sinusitis
|
|
|
neutropenia definition
|
decrease in absolute neutrophil count (ANC)
age related limits: term newborn <3000 infant <1100 child, adolescent, adult <900 altitude lower ANC |
|
|
neutropenia risk of infection
|
1000-1500 none
1000-500 minimal 200-500 moderate to severe <200 |
|
|
psuedo-neutropenia
|
low normal neutrophil count
treat with exercise or epinepherine |
|
|
infection induced neutropenia
|
common during viral infections (increased use)
|
|
|
drug induced neutropenia
|
immune mediated or direct
|
|
|
immune neutropenia
|
antibodies directed to neutrophils or their precursors
chronic benign neutropenia: occur in children and adults. infections are infrequent despite low ANC |
|
|
congenital neutropenia
|
cyclic neutropenia
autosomal disorder kostmann syndrome: severe congenital neutropenia |
|
|
lymphopenia
|
absolute lymphocyte count of <1000
decreased production increased destruction increased loss |
|
|
presentation of Acute myeloid leukemia
|
progressive fatigue, dyspnea,
CBC: WBC high normal, ANC - low nuetrophil hb low plts low |
|
|
acute myeloid dx
|
>20% blasts in marrow
|
|
|
potential side effect of Acute myeloid leukemia tx
|
Tumor lysis syndrome
rapid lysis of cells psills uric acide, into blood..causes kidney damage...phosphorous levels increase cardiac dysrythmia, calcium too low...causes nephropathy |
|
|
consolidation therapy for Acute myeloid leukemia
|
give high does of Ara-C for 12 hours at 1,3,5 days over 4 months
toxicity changes: neuropathy |
|
|
discharge drugs for Acute myeloid leukemia
|
acyclovir, fluconasol, moxyflocacil
|
|
|
good prognosis for acute myeloid leukemia
|
16.16 inversion
8-12 translocation |
|
|
bad prognosis for acute myeloid leukemia
|
deletion of 5 or 7
111q23 MDR |
|
|
Acute promyelocyte leukemia tx side effects
|
increased risk of DIC because primary granules formed in promyelocyte stage
(1)ATRA syndrome - fever, pulmonary tension (2)hyperleukocytosis |
|
|
Acute lymphocytic leukemia presentation
|
fatigue, leukopenia or leukocytosis, pallor, bruising, fever, hepatosplenomegaly
pancytopenia lymphadenopathy sepsis bleeding tumor lysis syndrome |
|
|
Tumor lysis syndrome released molecules
|
LDH
uric acid potassium phosphorous creatinine |
|
|
Components of ALL treatment
|
induction
consolidation CNS therapy - IV methotrexate do CNS therapy throughout all phases of treatment long term continuation |
|
|
good prognosis for Acute lymphocytic leukemia
|
<50k WBC
young age no CNS hyperdiploidy translocation 12.21 |
|
|
unfavorable prognosis for Acute lymphocytic leukemia
|
hypodiploidy
4.11 translocation 9.22 translocation (different protein product) |
|
|
presentation of chronic myelogenous leukemia
|
high WBC
increased neutrophils, bands, metamyelocytes, myelocytes, promyelocytes, and blasts high platelets |
|
|
presentation of chronic lymphocytic leukemia
|
symptoms of anemia or thrombocytopenia
immune dysregulation - prolonged infections hypogammaglobulinemia (low IgG) |
|
|
chronic myelogenous leukemia tx
|
gleevec - TKI binds to c-kit on GISTs
and PDGFR alpha |
|
|
why does gleevec work?
|
translocation of ABL (9.22) is always on
gleevec binds to site and prevents it from being on |
|
|
Immune symptoms of Chronic lymphocytic leukemia
|
anemia
immune dysregulation hypogammaglobulinemia over active destruction of self cells - RBCs and platelets (seen in later stages) |
|
|
treatment of chronic lymphocytic leukemia cos/benefit
|
early stages may just have high WBC, but not worth treatment until immune system dysregulation because of treatment costs
|
|
|
stages of chronic lymphocytic leukemia
|
0 - elevated lymphocyte count
1- elevated with lymphadenopathy 2 - elevated with splenomegaly 3 - elevated with anemia 4 - elevated with low platelets |
|
|
karyorrhexis
|
stage of cellular necrosis in which the fragments of the nucleus fragments and its chromatin are distributed irregularly throughout the cytoplasm.
associated with cobalamin/folate deficiency |
|
|
where is TPO produced?
|
liver
|
|
|
platelet function
|
(1) formation of mechanical plubs
(2) local release of vasoconstrictors (3) catalysis of reactions of the soluble coagulation cascade leading to fibrin clot formation (4) initiation of the tissue repair process (5) regulation of local inflammation |
|
|
heparin induced thrombocytopenia
|
IgG antibodies directed against the heparin-platelet factor 4 complex
suspect if platelet count falls to <100k venous, arterial, and microvascular thrombosis threatens life and limb |
|
|
pathogenesis heparin-induced thrombocytopenia
|
heparin binds to platelet factor 4
antibodies then bind to complex Fc receptor on platelets is stimulated by this antibody...platelets get rapidly consumed and a total decrease in available platelets |
|
|
Thrombocytopenia
|
<150k
Causes (1) decreased platelet production (2)decreased platelet survival (3) sequestration (4) dilutional |
|
|
Thrombocytosis
|
>500k
complications: (1)thrombotic episodes (2) bleeding Causes: (1) primary - essential thrombocytosis (2)secondary/reactive - infection, inflammation ,etc. |
|
|
Immune Thrombocytopenic Purpura
|
may be secondary or primary
(1)antiplatelet antibodies to GpIIb/IIIA or GpI dx: peripheral blood: thrombocytopenia bm: normal or increased mega's |
|
|
Coag Studies
Platelet Enumeration |
normal
150-350l |
|
|
Coag Studies
Bleeding time |
BT
normal 2-9 mins |
|
|
Coag studies
Aggregation studies |
addition of platelet aggregators - ADP
dx: platelet function tests |
|
|
coag studies
activated partial thromboplastin time (aPTT) |
normal is 21-25 second
dx: increased aPTT - sensitive to intrinsic pathway abnormalities XII, XI, IX, VIII, X, V heparin |
|
|
coag studies
prothrombin time (PT) |
normal 10-14 seconds
Dx: increased PT - sensitive to extrinsic pathway abnormalities: VII, X, V, II, fibrinogen useful in vitamin K deficiency (2,7,9,10) |
|
|
coag studies
Fibrinogen Degradation products (FDPs or D-dimers) |
measures presence of fibrin or fibrinogen degradation products
useful for: DIC - very elevated DVT - slightly elevated Problem: cannot distinguish between plasmin action on fibrinogen vs fibrin. Therefore can be elevated when there is no clot present. Usually: thrombin makes fibrin..fibrin makes 13, 13 cross links. only see D dimers when fibrin cross linked |
|
|
PT elevated
aPTT normal Platelets normal |
dx
7, 10, 5 deficiency, vit k deficiency |
|
|
PT normal
aPTT high Platelets norm |
8,9,11 def, VWD, heparin
|
|
|
PT high
aPTT high Platelets normal |
vit k def
liver disease |
|
|
PT high
aPTT high Platelets low |
DIC
|
|
|
PT norm
aPTT norm Platelets low |
hypersplenism, platelet destruction, platelet production problems, TTP, HUS
|
|
|
PT norm
aPTT norm Platelets high |
reactive vs MPD
|
|
|
PT norm
aPTT norm Platelets norm |
mild VWD
|
|
|
RBC fragmentation syndromes
|
(1) mechanical/vascular hemolysis
(2)microangiopathic hemolytic anemias (MAHA) DIC TTP Hemolytic uremic syndrome |
|
|
DIC - definition
|
a secondary condition in which microthrombi develop throughout the bloodstream, blocking small blood vessels and depleting platelts and clotting factors needed to control bleeding
|
|
|
broad terms DIC
|
simultaneous activation of
(1) coagulation system - widespread expression of TF a. intravascular thrombuc formation compromising blood supply to organs b. exhuastion of platelets and coagulation factors resulting in hemorrhage (2) fibrinolytic system - plasmin degrades fibrinogen and fibrin, producing D-dimers, exacerbating bleeding |
|
|
clinical pentad of TTP
|
(1)MAHA
(2)Neurologic abnormalities (3)renal insufficiency (4)fever (5) thrombocytopenia |
|
|
Laboratory Dx of TTP
|
(1)deficiency of vWF metalloproteinase ADAMTS13
(2) consumptive thrombocytopenia (3) schistocytes in blood smears |
|
|
Hemolytic Uremic Syndrome (HUS)
clinical features |
(1)Microangiopathic hemolytic anemia (MAHA)
(2)Thrombocytopenia (3)Renal insufficiency (4)bloody diarrhea |
|
|
Hemolytic Uremic Syndrome (HUS)
lab dx |
(1)E. coli infectious gastroenteritis
0157:H7 produces Shiga-like toxin (2)consumptive thrombocytopenia (3) schistocytes in blood smear |
|
|
von Willebrand's Factor
functions |
(1)carrier protein for VIII - increases serum half life
(2)binds to GpIb receptor on platelet adhesion to damaged endothelium |
|
|
von willebrand's disease
|
bleeding disorder - mucocutaneous bleeding disorders
auto dominant or recessive (1)mild form (type 1 and 2) - patients asymptomatic (2)severe forms (type 3) usually have factor 8 problems as well |
|
|
vWD dx
|
(1)hematologic eval of peripheral blood
(2)screening for hemostasis (3)vWD panel - multimeric analysis of vWF (a) Type 1 - generalized decrease (b) Type 2 - loss of HMW (c) Multimer absence |
|
|
Factor 8 deficiency
|
Factor 8 synthesized in liver, circulates bound to vWF, activated by IIa and enhances activation of 10 by 9a. Inactivated by Protein C
|
|
|
Hemophilia A
|
sex-linked recessive - Xq28
Factor 8 deficiency spectrum of bleeding manifestations suspect if: (1) family hx (2) appearance of bleeding in neonatal period (3) pattern of bleeding (joint, muscle) easy bruising |
|
|
Hemophilia B
|
factor 9 deficiency
vitamin K dependent activated by 11a and 7a X linked recessive or can be acquired with liver disease, vitamin K deficiency spectrum of bleeding disorders |
|
|
Acquired coagulopathies
|
(1)liver disease - major producer of all factors but vWF
(2)vitamin k deficiency - 2, 7, 9, 10, protein c/s (3) disseminated intravascular coagulopathy DIC |
|
|
Factor 5 leiden
|
most common inherited thrombotic disorder
auto dominant mutation in factor 5 renders the cofactor resistant to proteolysis by activated protein c recurrent venous thromboembolism Dx. DNA mutation analysis Tx. prophylaxis |
|
|
Anticoagulants/Fibrinolysis
|
(1)antithrombin III
downregulates 12a, 11a, 10a, 9a (2)proteins C and S downregulates 5a and 8a (3) Plasmin degrades fibrin making d-dimers |
|
|
Thrombosis
|
(1)endothelial injury
(2) abnormal blood flow (3) hypercoagulability |
|
|
Hypercoagulability
|
(1)primary - genetic thrombi
(2) secondary - arterial and venous |
|
|
Antiphospholipid Antibody Syndrome
|
prothrombotic disorder
(1) autoantibodies directed against a number of antigens complexed to phospholipids (2)recurrent venous and arterial thromboembolism, fetal loss, thrombocytopenia, neurologic manifestations (3) most often diagnoses because of incidental paradoxical elevated aPTT |
|
|
Hemophilia A genetics
|
Factor 8
X chromosome - long arm missense, frameshit, deletions or inversions inversion of intron 22 common, but most are point mutations |
|
|
Hemophilia A/B inheritance
|
sex linked recessive
30% spontaneous |
|
|
Clinical manifestations of Hemophilia A/B
|
joint bleeds - target joint where blood repeatedly leaks into
deep muscle bleeds hematomas dental extraction |
|
|
severity of hemophilia A/B
|
severe - present early childhood
moderate - after minor trauma or surgery mild - often unaware until trauma |
|
|
Complications of hemophilia
|
(1) hemophilic arthropathy - repeated bleeds cause synovitis, ultimately destroying synovial membrane and muscles
(2)synovitis and increased blood flow to the joint may lead to epiphyseal overgrowth and limb length discrepancy (3) intramuscular/soft tissue bleeding can cause compaction and ultimately extremity compartment syndrome |
|
|
Hemophilia B genetics
|
factor 9 deficiency
X linked mutation |
|
|
Lab findings for Hemophilia A
|
PFA normal
Platelet Count normal Prothrombin time normal (extrinsic pathway) activated partial thromboplastin time (aPTT) long (intrinsic pathway) |
|
|
Tx of hemophilia
|
prophylaxis
give missing factor if possible or FFP or plasma derivatives AMICAR - stabilizes fibrin clot |
|
|
Complications of hemophilia treatment
|
(1) development of inhibitors to factors
(2) infections |
|
|
Acquired hemophilia
|
development of neutralizing and clearing anti-factor 8 antibodies
more common in elderly or post partum |
|
|
von willebran disease genetics
|
auto dominant
bleeding due to reduced level or abnormal vWF |
|
|
vWF function
|
(1)promotes platelet adhesion to damaged endothelium
(2) carrier molecule for factor 8, increasing half life |
|
|
Clinical manifestations of VWD
|
mucous membranes - menses extremely common
skin cuts post trauma post op |
|
|
lab findings in VWD
|
PFA prolonged
platelet count normal PT normal aPTT normal/prolonged |
|
|
Tx of vWD
|
(1)DDAVP - stimulates release of factor 8 and vWF fro endothelial cells but only works for about 3 days
(2) Humate P - concentrated factor 8 and vWF |
|
|
Therapy based on levels of pain
|
mild pain - acetaminophen, NSAIDs, or mild opioids (darvocet)
moderate pain - small doses of oxycodone alone severe/debilitating pain - morphine |
|
|
Non-narcotic analgesics
|
Acetaminophen
Salicylates NSAIDs nonacetylated salicylates |
|
|
Acetaminophen use/benefits/metabolism
|
analgesic and antipyretic effects
metabolized in liver use with mild pain inhibits synthesis of prostaglandins in CNS and peripherally blocks brain impulse generation |
|
|
NSAIDs
|
analgesic, antipyretic, and anti-inflammatory effects
inhibits COX Dose response curve plateus particularly helpful in bone pain limitations - gastrophy, anti-platelet effect, renal toxicity, |
|
|
COX2 inhibitors
|
patients where an NSAID is indicated
history of GI ulcers elderly low platelet count receiving anti coagulation receiving corticosteroids (Celebrex) |
|
|
Celebrex
|
used for acute or chronic pain
use for FAP interacts with warfarin |
|
|
Tramadol
|
complex MOA
get benefit of opioids without addiction |
|
|
morphine
|
well tolerated
adverse: sedation, urinary retention, decreased respirations, constipation become tolerant to everything but constipation (must give with laxative) |
|
|
oxycodone
|
slightly more potent than morphine
milder side effect profile |
|
|
hydromorphone (dilaudid)
|
approximately 6 times more potent than morphine
less nausea/vomiting useful in liver/renal failure patients |
|
|
oxymorphone
|
very potent
only used by pain specialists |
|
|
fentanyl
|
available IV, PO, transdermal patch, transmucosal sucker
100x more potent than morphine heat increases absorption (fever and heat pads) |
|
|
meperidine
|
not recommended for use because of high toxicity
|
|
|
methadone
|
cheap form of pain management
dosing is complex opioid agonist and NMDA antagonist high drug interaction |
|
|
adjuvant therapy for pain
|
tricyclic antidepressants
anticonvulsants - neurotin corticosteroids - pain from infiltration |
|
|
Heparin MOA
|
MOA: complexes with AT3 to accelerate its ability to inactivate factors 2a, 9a, 10a
|
|
|
Unfractionated heparin tx
|
monitor aPTT when given as treatment
binds to AT3 to accelerate inactivation of factor 10a. it can also bind and inactivate thrombin. Measures of the intrinsic pathway...follow by aPTT |
|
|
Unfractionated heparin prophylaxis
|
do not need to monitor
binds to antithrombin to accerlate inactivation of factor 10a...can also bind and inactivate thrombin |
|
|
LMW heparin
|
do not monitor for tx or prophylaxis
|
|
|
Types of LMW heparin
|
enoxaparin - lovenox
dalteparin - fragmin fondaparinux - arixtra |
|
|
enoxaparin (lovenox)
|
indicated in the tx and prophylaxis of VTE
|
|
|
Dalteparin (Fragmin)
|
prevention of VTE and treatment of VTE
|
|
|
Fondaparinux
|
inhibits factor 10a
good option for herparin-induced thrombocytopenia (HIT VTE treatment and prophylaxis can cause renal insufficiency |
|
|
warfarin MOA
|
inhibits the production of vitamin k dependent factors: 2,7,9,10
protein C, protein S |
|
|
warfarin
|
monitor INR - varies by indication
most slowly adjust rates because warfarin can readily vary by interactions (1) highly protein bound so affected by other drugs (2) vitamin k levels vary by diet - leafy green veggies monitor PT (prothombin time) extrinsic pathway |
|
|
Aspirin
|
MOA: inhibits cyclooxygenase which blocks the formation of (1) thromboxane-TXA2---inhibits platelet aggregation and activation
(2) prostacyclin |
|
|
Ticlopidine
|
MOA: inhibits platelet activation and aggregation
interacts with CYP3A4 |
|
|
Clopidogrel
|
MOA: blocks ADP receptors---inhibits platelet activation and aggregation
prevents activation of GP2b/3a |
|
|
heparin induced thrombocytopenia (HIT)
|
heparin binds to platelet factor 4
an IgG antibody binds to the heparin/PF4 complex. This complex binds to an activated platelet surface. Results in platelet clearance (thrombocytopenia) |
|
|
Heparin-induced thrombocytoepnic thrombosis (HITT)
|
heparin binds to platelet factor 4. This complex binds to an activated platelet surface. An IgG antibody binds to the heparin/PF4 complex. Results in platelet clearance - get thrombocytopenia.
Fc portion of IgG further activates platelets, resulting in thrombosis. |
|
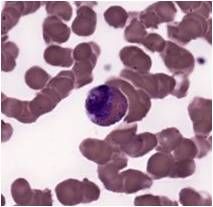
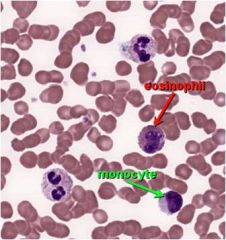
|
eosinophil
|
|
|
|
|
|
|
|
|
|
|
|
basophillic erythroblasts
|
|
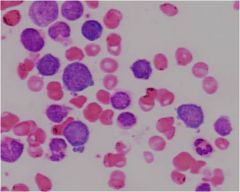
|
basophillic on the left
polychromatic center and right |
|
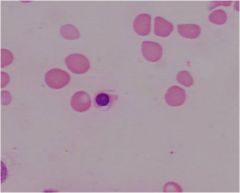
|
orthochromatophillic erythroblast
|
|
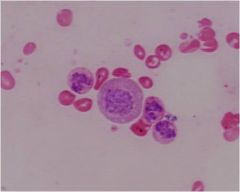
|
promyelocyte
|
|
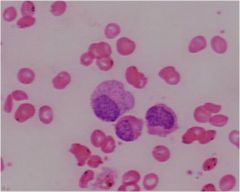
|
myelocyte
|
|
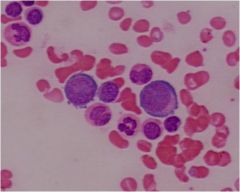
|
metamyelocyte with indenting nucleus
|
|
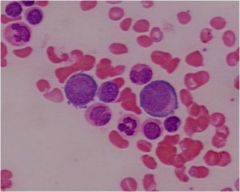
|
neutrophil
|
|
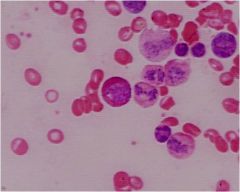
|
eosinophilic myelocyte
|
|
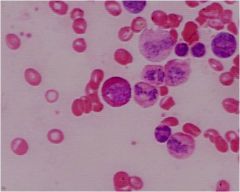
|
eosinophilic myelocyte
|
|
|
eosinophillic metamyelocyte
|
|
|
|
|
|
|
|
|
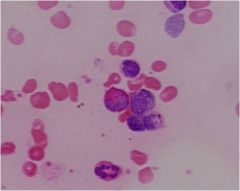
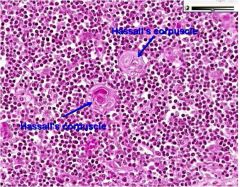
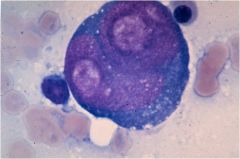
Reed Sternberg
|
|
|
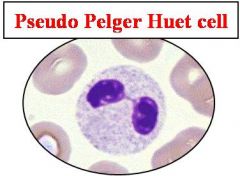
pseudo pelger huet cell
associated with MDS |
|
|
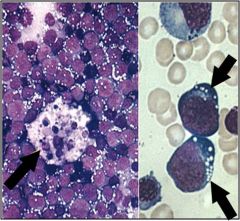
Burkitt's Lymphoma
|
|
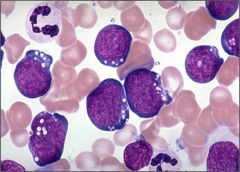
|
burkitt's lymphoma
|
|
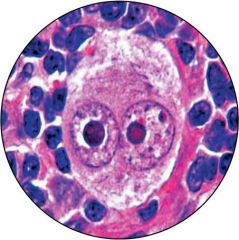
|
Reed Sternberg
Classical hodgkins |
|
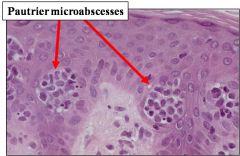
|
Pautrier microabscesses associated with mycosis fungoides
|
|
|
Parvovirus B19 clinical syndromes
|
(1) hydrops fetalis
(2) erythema infectiosum (fifth diease) (3) aplastic crises in hemolytic subjects (4) seronegative RA syndrome - Arthropathy (5) chronic anemia in immunocompromised hosts |
|
|
Parvovirus B19 pathogenesis
|
infects EPC
acute infection causes mild illness in most immunocompetent children rash about time of seroconversion fever, myalgias, HA precede rash by a few days |
|
|
EBV pathogenesis
|
tropism for monocytes
|
|
|
Phase I engraftement
|
pre-engraftment - neutropenic
most likely to get G-, staphylococcus, GI strepp |
|
|
Phase II post-engraftment
|
impaired cell mediated immunity
get viruses CMV, EBV, |
|
|
Phase III engraftment
|
impaired cellular and humoral immunity as B cells maturation takes a few years
more likely to get encapsulated organism infections (Pneumo) |
|
|
Risk factors of CMV in BMT recipients
|
seropositive status
older age conditioning regimens with agents other than cyclophosphamide GvHD |
|
|
Therapy for CMV
|
IV Gamcyclovir+ Ig either CMV or IVIg
Gamcyclovir is myelosuppressive - target prophylaxis recommended |
|
|
Strategies to reduce risk of CMV disease
|
(1) prophylaxis
(2) preemptive therapy for targeted patients |
|
|
Herpes Zoster incidence with age
|
with increasing age, increasing incidence
|
|
|
Vaccine for Herpes Zoster
|
helps reduce risk in older adults in healthy adults
but no vaccine approved for immunocompromised |
|
|
At what stage of granulocyte development are the specific granules that differentiate the cell types first apparent?
|
Specific granules first appear at the myelocyte stage.
|
|
|
– In many cases clusters of developing blood cells comprise predominantly white cells or red cells, rather than highly-intermixed groups of both. What accounts for this relative segregation?
|
Because of the progressive restriction of the potentiality of blood precursor cells, many colonies form which will produce only one type of blood cell (e.g., erythrocytes, granulocytes or monocytes). Remember also that growth factors, which may act over some distance, influence the developmental pathways. Finally, the developing cells continue to divide through the earlier stages, so that also contributes to the clustering of cells in the same lineage.
|
|
|
What accounts for the basophilia of the cytoplasm in early stages in erythrocyte development and the eosinophilia in later stages?
|
In the early stages of red cell development the large number of polyribosomes required for the production of hemoglobin result in basophilic staining in the cytoplasm (because of the basophilia of the RNA associated with the polyribosomes). As hemoglobin accumulates, eosinophilia in the cytoplasm increases and basophilia decreases as the number of polyribosomes declines.
|
|
|
– In hereditary spherocytosis the normal binding between the proteins ankyrin and spectrin is absent. What type(s) of blood cells would be affected in this disorder? In what way(s) might their structure and/or function be compromised and why?
|
Normally, the internal surface of the cell membrane of erythrocytes is braced by cytoskeletal proteins through interactions of the proteins ankyrin and spectrin. When the normal binding of those proteins is compromised, the red cell membrane is not braced and the cell is easily deformed and does not form its normal, biconcave shape. The cells are more fragile and do not resist changes in osmotic pressure. The cells are more vulnerable to sequestration and destruction by macrophages.
|
|
|
What obvious cytological feature of RBCs accounts, in large part, for their limited life span? In what organ(s) are worn-out or damaged RBCs destroyed?
|
Mature erythrocytes do not have a nucleus or other organelles necessary for protein synthesis and thus no ability to replace worn-out or damaged structural proteins and enzymes. Worn out erythrocytes are removed primarily by macrophages in the spleen and bone marrow (also in the liver, I believe).
|
|
|
What type(s) of cytoplasmic granules are common to all granulocytes? What type(s) differ among the granulocytes? How does this relate to the cells’ functions?
|
All granulocytes contain azurophilic granules (aka primary granules), which are essentially lysosomes (even the agranulocytes, lymphocytes and monocytes, may show some azurophilic granules). These granules contain acid hydrolases, as well as antibacterial and digestive substances. The specific, or secondary granules, differ among the three types of granulocytes. The specific granules in each type of granulocyte contain a variety of substances, some of which are easily related to the specific functions of each cell type. For example, lysozyme in the neutrophilic granules is involved in digesting phagocytosed bacteria, the major basic protein in eosinophilic granules appears to function in the killing of parasites, and basophilic granules contain heparin and histamine, which play a role in the early inflammatory response and allergic reactions.
|