![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
71 Cards in this Set
- Front
- Back
|
What is the viscosity of blood? |
1.1-1.2 Centipoise |
|
|
What are the factors that influence blood viscosity |
RBC and protein conc.
increased cells increase viscosity
smaller vessels increase viscosity |
|
|
What is the average volume of blood in a person? |
70mL/kg for adults
children may have slightly higher. ~10mL/kg |
|
|
What is aperture impedance? |
Based on the detection of electrical resistance as cells pass through a small aperture.
Voltage pulses determine number of cells Height of pulses determine cell volume |
|
|
how does light scatter and fluorescence measure cells |
degree of scatter: 0= size 7= complexity 90= lobularity 90 (d)= granularity |
|
|
What is RDW |
Red cell distribution width. can measure anisocytosis |
|
|
What is the lifespan of cells in the blood? |
RBCs 120 days Platlets 10 days Granulocytes hours
|
|
|
Name the Haematopoietic tissue |
Fetus 0-2 months = yolk sac 2-7 months = liver/spleen 5-9 months = bone marrow
infants = bone marrow (all bones)
Adults = vertebrae, ribs, sternum, skull, sacrum and pelvis, proximal end of femurs |
|
|
Haemopoiettic Stem cells |
~1% of bone marrow CD34 expressed Pluripotent Can self renew |
|
|
Describe the haemopoietic cell % in bone marrow |
Erythropoietic cells ~20% Granulopoetic cells ~60% Lymphocytes ~15% Megakaryocytes <0.1% Other: Plasma and macrophages |
|
|
WBC diff %s |
Neutrophils 40-75% Eosinophils 1-5% Basophils <1% Monocytes 2-8% Lymphocytes 20-45% |
|
|
Platlet range |
150-400 x10^9/L |
|
|
Reticulocyte description |
Anuclear Spherical shape mRNA remains (bluish colour) polychromataphilia Can be found in blood |
|
|
Haemoglobin structure |
A A2B2 (A from C16, B from C11) A2 A2D2 (D from C11) F A2G2 (G from C11) |
|
|
Hb prevelence |
Hb A 95% in adult HbF small % after birth HbA2 small levels are normal in adults |
|
|
Unloading vs Loading of 02 |
Basic and low 2,3 DPG favors loading
Acidic and high 2,3 DPG favors unloading |
|
|
RBC membrane composistion |
Lipid Bi layer Spectrin, actin, arkyrin and Band 3 |
|
|
RBC metabolism |
Glycolysis, (anaerobic production of ATP)
Hexose monophosphate shunt keeps GSH in a reduced state via NADP->NADPH
methaemoglobin pathway keeps Fe in a 2+ state
|
|
|
Folate and B12 |
Required for DNA production
daily requirments is: 1ug for b12 100ug for folate |
|
|
Reference range |
WBC 4-10 x10^9/L Hb 115-160 g/L Plt 150-400 x10^9/L RBC 3.9-5.6 x10^12/L Hct .38-.45 MCV 800-100fL RDW 9-15 MCH 27-32pg MCHC 320-360 g/L
|
|
|
Causes of Iron deficiency |
Chronic blood loss
Malabsorbtion
Poor diet
Phlebotomy |
|
|
Iron deficient anaemia symptoms |
Shortness of breath tiredness headaches pallor of the mucous membranes tachycardia
|
|
|
Causes of macrocytosis |
Alchol, liver disease, pregnancy, hyperthyroidism, reticulocytosis, aplastic anaemia, red cell aplasia, folate and b12 def,myeloidsplasia and paraproteinaemia
|
|
|
macrocytic anaemia |
Tear drop cells ovalmacrocytes hypersegmented neutrophils
|
|
|
Microcytic anaemia |
Tear drop cells target cells pencil cells
|
|
|
α-Thalassaemia types |
αα/α- silent/thalassaemia trait αα/-- or α-/α- thalassaemia trait α-/-- Hb H disease --/-- Hb Barts/ Hyrops fetalis |
|
|
Hb barts / Hardrops fetalis |
Almost all Hb is Hb H total failure of the α-globin gene most die during pregnancy or die after birth |
|
|
Hb H disease |
Greatly reduced α-globin synthesis results in hypochromatic microcytic anaemia Excess ß-globin results in Hb H (ß4) Hb H can precipitate anc cause RBC lyses |
|
|
Clinical Features of Hb H |
May be mild Hb ussually 70-100g/l MCV 50-65 MCH 15-20 Spleenomegaly Could have jaundice
|
|
|
α-thalassaemia trait |
Asymptomatic Microcytosis W/ or W/out anaemia Partner testing if patient is αα/-- |
|
|
ß-thalassaemia |
Results from reduced rate of ß-globin most common are point mutations in ß-globin locus >200 types reported |
|
|
Categories of ß-thalassaemia |
ß naught- No globin expression from the gene
ß+ - Reduced production of ß-globin |
|
|
clinical types of ß-thalassaemia |
ß- major ß- intermedia ß- Trait |
|
|
ß-thalassaemia trait |
Heterozygosity for ß thalassaemia usually asymptomatic may become symptomatic when under stress
|
|
|
ß-thalassaemia trait clinical indicies |
↓MCV/MCH ↑RCC Hb studies: ↑Hb A2 +/- ↑Hb F |
|
|
ß-thalassaemia major |
Homozygous or compound heterozygous for ߺ thalassaemia excess α-globin precipitates and causes damage to developing RBCs ↓RBC life Transfusion dependent anaemia Not present inu tero or at birth Presents after 1st year of life |
|
|
Symptoms of ß-thalassaemia major |
Bony deformaties hepatosplenomegaly transfusion dependent after 1st year of life growth of sexual organs can be delayed survival and quality of life is transfusion dependent BM transplant is curative but there is sig mortality |
|
|
ß-thalassaemia clinical indices |
↓↓MCV/MCH Hb Studies: Hbf and Hb A2; minimal to no Hb A |
|
|
ß-thalassaemia intermedia |
Anaemia hb 70-100g/l may have spleenomegaly and bony deformities Not usually transfusion dependant
Types ߺ ß+ ß+ ß+ ßn ߺ(dormant) ßn ߺ +excess α chains (ααα) |
|
|
Delta Beta thalassaemia |
Deletions of ß locus which removes both ß, δ genes similar clinical features as ߺ thalassaemia hypochromatic microcytic ±anaemia ↑HbF Hb A2 is low or normal |
|
|
Hb Lepore |
Fusion between ß and δ genes similar to ß trait Partner testing should be offered
|
|
|
Hb E |
Substitution mutation Hb E trait asymptomatic, 50% will have abnormal indices HB E disease usually asymptomatic mild anaemia with microcytosis
|
|
|
HbE/ß thalassaemia |
variable severity (trait, major, intermedia) not easy to predict phenotype ~50% will be transfussion dependant ~50 will require splenectomy |
|
|
Hb C |
ß-globin varient common in africa Heterozygous Hb C is asympotmatic Homo Hb C: Spleenomegaly and gallstones may be present Significant when co-inherited with Hb S |
|
|
Hb S |
Glu>Val replacement deoxy Hb S undergoes polymerisation When there is enough Hb S polymer there will be Sickling will cause sickle cell disease
|
|
|
Sickle cell disease types |
Homo Hb SS Hetero: Hb S/C, Hb S/ß thalassaemia, Hb S/D punjab, Hb S/C harlem Hb S/O arab
|
|
|
Describe iron overload |
Raised serum iron and transferrin (>45%) Raised serum ferritin increased iron in the liver and heart abnormal liver function tests
|
|
|
organ dysfunction with iron overload |
Cardiomyopathy reduced sexual organ development Diabetes Hyperthyroidism cirrhosis, haemosiderosis
|
|
|
Iron overload pathogenesis |
Genetic haemochromatosis 95% homo for HFE mutation
HFE chromosone 6 (90% for c282y mutation) Juvenile haemochromatosis (HFE2) Chromosone 1 Haemochromatosis type 3 HFE3 classic phenotype but no HFE mutation
African Iron overload Excess iron intake ineffective erythropoiesis Repeated blood transfusion
|
|
|
Intrinsic pathway |
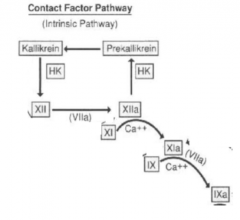
|
|
|
extrinsic pathway |
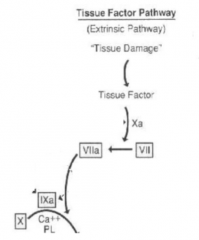
|
|
|
common pathway |
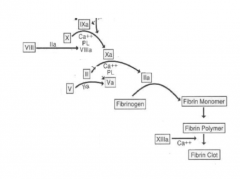
|
|
|
inhibition of clotting cascade |
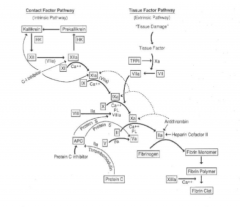
|
|
|
Finrinolyses |
FIIa stimulates the tPA, uPA is released from endothelial cells. tPA and uPA activate plasminogen to plasmin Plasmin breaks down fibrin |
|
|
Haemophilia A |
80-85% of Haemophilias prolonged aPPT and not PT Plasma FVIII ~1% Treated with recom FVIII |
|
|
Haemophilia B |
Prolonged aPTT and Normal PT time ↓Factor IX Treated with conctrated F IX |
|
|
lupus anticoagulant |
Antibodies bind to phospholipids, this reduces the amount in plasma, thus increases the aPTT, dRVVT and PT. Adding excess phospho lipids correct it. Mixing studies does not correct |
|
|
vWF |
Encoded on chromosome 12 Produced in endothelial cells and in MK's mulitmers range from 1000-20000kDa
|
|
|
Functions of vWF |
Binds to collagen which links platlets stabilises fVIII |
|
|
vWF disease |
Type 1: 70% if vWD normal functioning vWF but in redused amounts
Type 2: functional deficiency of vWF
Type 3: No detectable vWF |
|
|
coag and pregnancy |
↑fibrinogen, II, V, VII, VIII, IX, and X
↓XI, XIII, fibrinolysis and protein C and S.
|
|
|
schistocytes and their causes |
Mechanical damage to the microvascular (abnormal surfaces, cardiac lesion, fibrin strands) Can for micro spherocytes usually reticulocytes present sharp pointy ends assoc with thrombocytopaenia (disseminated intravascular coag, thrombotic thrombocytopaenic purpura, Haemolytic ureamic syndrome.)
|
|
|
Haemolysis from infections |
direct damage to RBC (malaria) Toxin production (clostridium) Oxidant stress microangiopathic HA (meninoccaemia) Auto antibody prod |
|
|
Enviromental |
KIdney disease: burr cells Hepatic dysfunction: Acanthocytes |
|
|
Lab features of haemolysis |
Hb Norm or ↓ Reticulorcytes ↑ polychromasia spherocytes, fragmented cells ↑uncojugated bilirubin BM: erythroid hyperplasia |
|
|
Intrinsic Red cells defects |
Hereditory spherocytosis enzyme defect g6pf Thalassaemia abnormal red cells
|
|
|
Extrinisic red cell defects |
Severe hepatic or renal dys red cell frag: DIC, HUS, TTP Infections Auto or allo-immune
|
|
|
Heriditory spherocytosis |
Defect of RBC membrane proteind (Band 3, spectrin, actin) most testing targets band 3, Fluctuating anaemia and jaundice RBCs prematurely destroyed in spleen spleenomegaly and gallstones |
|
|
Diagnosis of heriditory spherocytosis |
hb can be variable blood film; spherocytes and polychromasia flow cyt EMA shows reduced binding to band 3 direct anglobulin test |
|
|
Diagnosis of spherocytes |
heriditory spherocytosis auto immune HA drug induces HA ABO HA
|
|
|
Megoblastic anaemia |
Inhib of DNA but not RNA in ↓b12 deficiency there is an inhability to use n-Methy-tetrahydrofolate In ↓Folate deficiency there is an inhability to synthesise n-methy-tetrahydrofolate → ↓ thyamine for DNA synth |