![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
138 Cards in this Set
- Front
- Back
|
Chromatin Structure: DNA exists in the condensed, chromatin form in order to fit into the nucleus. How many times does negatively charged DNA loop around a positively charged histone?
What is this structure called? |
twice
nucleosome = "bead"
Think of beads on a string! |
|
|
What amino acids are histones rich in? |
lysine and arginine |
|
|
Which type of Histone has a role in binding to BOTH the nucleosome and to the "linker DNA"?
What is the function of this type of histone?
Which type(s) of histone are found in the nucleosome core? |
H1 (the only histone not found in the nucleosome core)
stabilizes chromatin fiber
H2A, H2B, H3, H4 (each 2x) |
|
|
Antihistone antibodies are found in patients with drug-induced lupus. What medications are commonly associated with drug-induced lupus. |
It's not HIPP to have lupus.
Hydralazine Isoniazid Procainamide Phenytoin |
|
|
In mitosis, DNA condenses to form ____________.
DNA and histone synthesis occur during ____ phase. |
chromosomes
S |
|
|
Define Heterochromatin. |
condensed, transcriptionally inactive, sterically inaccessible
HeteroChromatin = Highly Condensed |
|
|
Define Euchromatin. |
Less condensed, transcriptionally active, sterically accessible
Eu = true, "truly transcribed" |
|
|
During PROkaryotic DNA replication and the functioning of mismatch repair enzymes, how is the template strand distinguished from daughter strand? |
DNA methylation of cytosine and adenine found on the template strand |
|
|
How is transcription of CpG islands repressed? |
DNA methylation
CpG Methylation Makes DNA Mute |
|
|
How does histone methylation affect DNA transcription? |
Usually reversibly represses DNA transcription, but can activate it in some cases.
Histone Methylation Mostly Makes DNA Mute |
|
|
How does histone acetylation affect DNA transcription? |
Relaxes DNA coiling, allow for transcription.
Histone Acetylation makes DNA Active. |
|
|
Nucleotides: Which nucleotides are considered Purines?
Which nucleotides are considered Pyrimidines?
How many rings with each type? |
A, G = PURe As Gold
C, U, T = CUT the PY
Purines = 2 rings Pyrimidines = 1 ring
|
|
|
Nucleotides: Which nucleotide has a methyl?
What does deamination of cytosine result in? |
Thymine has a methyl
uracil |
|
|
State the number of hydrogen bonds found in G-C bond compared to A-T bond.
How does this affect the melting temperature of DNA? |
G-C bond = 3 H bonds A-T bond = 2 H bonds
increase G-C content = increase melting temperature of DNA |
|
|
What amino acids are necessary for the synthesis of purines? |
GAG Glycine Aspartate Glutamine |
|
|
Draw structure of purine and pyrimidine.
Which parts of the structure stem from amino acids and other sources? |
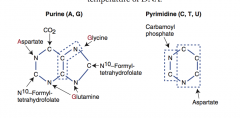
|
|
|
Differentiate Nucleoside from Nucleotide. |
NucleoSide = base + (deoxy)ribose (Sugar)
NucleoTide = base + (deoxy)ribose + phosphaTe; linked by 3'-5' phosphodiester bond |
|
|
Draw the pathway of pyrimidine and purine synthesis (de novo) |
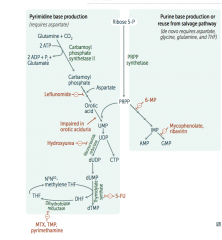
|
|
|
Describe the de novo production of AMP and GMP of the purine pathway: beginning with ribose-5-phosphate.
Describe elements/drugs that inhibit various aspects of the pathway?
What is rate limiting step? |
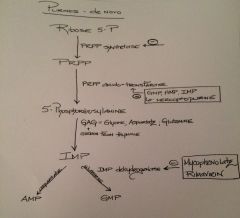
Rate limiting: PRPP amido-transferase |
|
|
Describe the de novo production of CTP and dTMP of the pyrimidine pathway: beginning with ribose-5-phosphate and production of carbonyl phosphate
Describe elements/drugs that inhibit various aspects of the pathway?
What is rate limiting step?
|
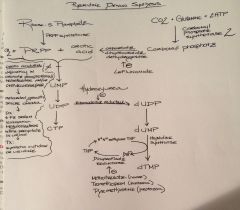
Rate limiting: CPS2 |
|
|
Describe the pathology of Orotic Aciduria. |
Pathology: Autosomal recessive inability to convert orotic acid to UMP (de novo pyrimidine synthesis pathway) because of defect in UMP synthase Note--UMP synthase is a bifunctional enzyme = orotate phosphoribosyl transferase and OMP decarboxylase
|
|
|
Describe the clinical findings of Orotic Aciduria. |
Findings: increase in orotic acid in urine ( white precipitate) megaloblastic anemia (does not improve with administration of B12 and folic acid) failure to thrive
ddx: no hyperammonemia compared to OTC deficiency, which does have increase orotic acid with hyperammonemia and decreased BUN
|
|
|
Describe the treatment of Orotic Aciduria |
oral uridine administration |
|
|
Draw the purine salvage pathway. |
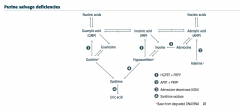
|
|
|
Give examples of some various compounds where the purine salvage pathway can be derived from? |
high energy compounds (ATP, DNA, RNA, GTP, etc) |
|
|
Describe the pathology of Adenosine deaminase deficiency. |
autosomal recessive defect of adenosine deaminase results in a build up of high energy compounds (i.e. ATP and dATP). ATP and dATP cause feedback inhibition of ribonucleotide reductase. Limited activity of ribonucleotide reductase will prevent DNA synthesis and thus decrease lymphocyte count.
One of the major causes of SCID Since developing T cells and B cells are some of the most mitotically active cells, they are highly susceptible to this condition. SCID happens to Kids. |
|
|
What is the function of ribonucleotide reductase? |
converts ribonucleotides to deoxyribonucleotides (basic building blocks of RNA to basic building blocks of DNA) |
|
|
What is the first disease to be treated by gene therapy? |
Severe Combined Immunodeficiency Disease
--note: absence of thymus in XRAY no plasma cells; B or T cells on CBC
Children have recurrent infections, but especially with Candida and Pneumocystis jiroveci |
|
|
What is the function of HGPRT? |
converts hypoxanthine to IMP converts guanine to GMP (resulting in salvage of purines) |
|
|
Describe the pathology of Lesch-Nyhan Syndrome. |
X-linked recessive absence of HGPRT results in defective purine salvage
causes: excess uric acid production and de novo purine synthesis (keep in mind, this will increase both IMP and PRPP, some of the output of PRPP will result in an increase in pyrimidine de novo synthesis too) |
|
|
Describe the clinical features of Lesch-Nyhan Syndrome. |
Findings: intellectual disability, self-mutilation, aggression, hyperuricemia, gout, dystonia.
HGPRT: Hyperuricemia Gout Pissed off (aggression; self-mutilation) Retardation (intellectual disability) DysTonia |
|
|
Describe the treatment of Lesch-Nyhan Syndrome. |
Xanthine oxidase inhibitors: allopurinol febuxostat (2nd line) |
|
|
Genetic Code features: Define unambiguous. |
each codon specifies only 1 amino acid |
|
|
Genetic Code features: Define degenerate/redundant |
most amino acids are coded by multiple codons Exceptions: methionine (AUG) and tryptophan (UGG) encoded by only 1 codon |
|
|
Genetic Code features: Define Commaless, nonoverlapping |
read from a fixed starting point as a continuous sequence of bases Exception: some viruses |
|
|
Genetic Code features: Define Universal. |
Genetic code is conserved throughout evolution Exception: In humans, mitochondria |
|
|
What features does both prokaryotes and eukaryotes share in the process of DNA replication? |
semiconservative and involves both continuous and discontinuous (Okazaki fragment) synthesis |
|
|
Describe the origin of replication. |
Particular consensus sequence of base pairs in genome where DNA replication begins. May be single (prokaryotes) or multiple (eukaryotes). |
|
|
Describe the replication fork. |
Y shaped region along DNA template where leading and lagging strands are synthesized.
Leading strand synthesized in the direction of which the replication fork is moving. Lagging strand synthesized in opposite direction of which the replication fork is moving. |
|
|
What is the function of Helicase? |
Unwinds DNA template at replication fork. |
|
|
What is the function of single-stranded binding proteins? |
prevents strands from reannealing. |
|
|
What is the function of DNA topoisomerase? |
Create a single or double stranded break in the helix to add or remove supercoils. (regulates overwinding or under-winding of DNA) |
|
|
What is the function of gyrase? |
is an enzyme that relieves strain while double-strand DNA is being unwound by helicase
found in prokaryotes, some eukaryotes, but not humans, which makes it a good target for antibiotics |
|
|
In regard to drugs targeting DNA replicating enzymes found in prokaryotes, what is the function of fluroquinolones? |
inhibits DNA gyrase inhibits prokaryotic topoisomerase II |
|
|
What is the function of Primase? |
makes an RNA primer on which DNA polymerase III can initiate replication. |
|
|
What is the function of DNA polymerase III? |
Prokaryotic only
elongates leading strand by adding deoxynucleotides (dNTPs) to the 3'-hydroxyl end. elongates lagging strand until it reaches primer of preceding fragment
3' to 5' exonuclease activity "proofreads" each added nucleotide.
DNA polymerase III has 5' to 3' synthesis and proofreads with 3' to 5' exonuclease.
|
|
|
What is the function of DNA polymerase I? |
Prokaryotic only Degrades RNA primer; replaces it with DNA.
Has same function as DNA polymerase III, but also excises RNA primer with 5' to 3' exonuclease. |
|
|
Contrast sites of replication between eukaryotes and prokaryotes. |
Prokaryotes: one one site (rich in AT base pairs) bound to DnaA proteins NOTE: DnaA proteins = bind to origin of replication and cause dsDNA to become a local region of ssDNA
Eukaryotes: multiple sites that include a short sequence that is rich in AT base pairs (consensus sequence) |
|
|
What is the function of DNA ligase? |
Seals = catalyzes formation of phosphodiester bond within a strand of double stranded DNA Example: Joins Okazaki fragments
|
|
|
What is the function of telomerase?
|
An RNA-dependent DNA polymerase adds DNA to 3' end of chromosomes to avoid loss of genetic material with each duplication |
|
|
What is a silent mutation? |
nucleotide substitution, but codes for same amino acid; often base change in 3rd position of codon (tRNA wobble) |
|
|
What is a missense mutation? |
nucleotide substitution resulting in changed amino acid (called conservative if new amino acid is similar in chemical structure) Seen in Sickle cell disease |
|
|
What is a nonsense mutation? |
nucleotide substitution resulting in early STOP codon.
STOP the NONSENSE! |
|
|
What is a frameshift mutation? |
deletion or insertion of a number of nucleotides no divisible by 3, resulting in a misreading of all nucleotides downstream, usually resulting in a truncated, nonfunctional protein
Seen in: duchenne muscular dystrophy |
|
|
Mutations in DNA -- Severity of damage: |
silent << missense < nonsense < frameshift |
|
|
For silent, missense and nonsense mutations (point mutations): Describe Transition and transversion. |
Transition: purine to purine or pyrimidine to pyrimidine Transversion: purine to pyrimidine or pyrimidine to purine |
|
|
What is nucleotide excision repair? |
Specific endonucleases release the oligonucleotide-containing damaged based; DNA polymerase and ligase fill and reseal the gap, respectively. Repairs bulky helix-distorting lesions |
|
|
In what disease process is nucleotide excision repair defective? |
xeroderma pigmentosum = this prevents repair of pyrimidine dimers due to UV light exposure
autosomal recessive mutations in XP repair genes (XPA through XPG) higher frequency in Japan defect in UV-specific endonuclease (aka. uvrABC excinuclease)
In healthy individuals, damage is excised by endonucleases; DNA polymerase replaces missing base pairs and ligase will seal it.
Also defective in (see future cards): Cockayne syndrome Trichothiodystrophy |
|
|
What is base excision repair? |
Base-specific glycosylase recognizes altered base and creates AP site (aka. a basic site or apurinic/apyrimidinic site). One or more nucleotides are removed by AP-endonuclease, which cleaves the 5' end. Lyase cleaves the 3' end. DNA polymerase-beta fills the gap and DNA ligase seals it. |
|
|
What is an AP site? When do they occur? |
also known as apurinic/apyrimidinic site or abasic site location where there is not a purine or a pyrimidine due to DNA damage (toxic deamination) or arises spontaneously |
|
|
What is the difference in the extent of repair between nucleotide excision repair and base excision repair? |
base excision repair and nucleotide excision repair are relatively similar, except base excision repair is only 1 altered based compared to nucleotide excision repair correcting longer stretches of nucleotides (2-30 bases) |
|
|
What is mismatch repair? |
Utilized when there is an error in the pairing of nucleotides secondary to DNA replication or recombination. The base pair mismatch repair system detects errors that escaped proofreading during DNA replication.
Identify the mismatched strand (note: methylated strand is parent DNA strand compared to unmethylated daughter strand--adenine residues in GATC sequence motifs are methylated)
Repair damaged DNA: mismatched nucleotides are removed, and the gap is filled and resealed.
|
|
|
In what pathology is mismatch repair defective? |
Hereditary nonpolyposis colorectal cancer (HNPCC)
Genes: MSH2 MLH1 |
|
|
What is a double strand break? What two processes can repair double strand breaks? |
both strands of the DNA double helix are broken. In this case, no template exists to guide the cell's repair process.
homologous recombination (recombinatorial repair or crossing over) non homologous end joining |
|
|
What is non homologous end joining? |
Repairs double strand breaks NOT during replication!
The cell attempts to repair the double strand break because it is highly deleterious for the cell: DNA ligase containing complexes join the separated ends of the double helix, relying on micro homologies between the ends of the single stranded fragments
This means: NHEJ is always mutagenic! |
|
|
What is recombinatorial repair? |
Repairs double strand breaks DURING replication!
A fragment of DNA has already been replaced and can serve as a template for the repair of the double strand break.
Mechanism similar to chromosomal crossover (nucleotide sequences are exchanged between two similar or identical molecules of DNA) |
|
|
What is translesion synthesis and when is it used? |
As a last resort, cells perform translesion synthesis as a means to continue DNA replication (when DNA damage is extensive enough to prevent replication machinery from advancing).
Special DNA polymerases repair damaged DNA by inserting nucleotide bases--this induces mutations by necessity in order to continue DNA replication. |
|
|
What are the clinical features of Xeroderma Pigmentosum? List the different stages. |
Patient's born with normal skin. First signs become evident early in life (by 6 mo of age)
First signs: freckle-like increased pigmentation and diffuse erythema and scaling, especially in light exposed areas
2nd stage of dz: development of telangiectasias, skin atrophy, mottled irregular pigmentation and or characteristic poikiloderma
Final stage: malignancies (i.e malignant melanoma; squamous and basal cell carcinomas; fibrosarcoma)
In addition, patients exhibit generalized photophobia and conjunctivitis (80%) Neurologic problems (20%): microcephaly, spasticity, hyporeflexia, ataxia, motor neuron signs and mental retardation |
|
|
Treatment of Xeroderma Pigmentosum: |
avoid sunlight
oral retinoids to reduce skin cancer, but they cause irreversible calcification of tendons and ligaments
5-FU and topical imiquimod and acitretin used to treat keratosis |
|
|
What are the cellular characteristics and genetics found in Fanconi Anemia? |
At least 11 genes involved and renders cells to be more susceptible to damage by free-O2 radicals. These mutations also cause deficiencies in DNA repair mechanisms and interfere with cell cycle control.
risk of malignancy is increased Hematopoietic cells are particularly affected (the majority develop cancer, particularly AML, and have bone marrow failure)
autosomal recessive higher incidence in Ashkenazi Jews and Afrikaners
|
|
|
What are the clinical features of Fanconi Anemia? |
DNA repair defects will cause:
bone marrow failure (pancytopenia as a consequence of aplastic anemia; leukemias and solid tumors) bone marrow failure symptoms: petechiae; bruising; pallor; fatigue; infections
Common cancers: liver, head and neck, esophageal and vulvar cancers
Newborns: genitourinary problems and poor growth
Pigmentation and cafe-au-lait spots |
|
|
Treatment of Fanconi Anemia: |
treatment focused on bone marrow failure, leukemias and solid cancers
keep in mind: using cancer drugs in patients with patients that have DNA repair defects can result in further complications for healthy cells |
|
|
What are the genetic and cellular characteristics in Cockayne syndrome? |
autosomal recessive mutations that result in faulty nucleotide excision repair of cells damaged by exposure to UV light
results in accumulation of mutations and overall accelerated aging of the cells
However, unlike XP, skin malignancies are UNCOMMON |
|
|
What are the clinical features of Cockayne syndrome? |
bird like face: large ears, thin nose, microcephaly with deeply set eyes
short stature (dwarfism) and long limbs
progressive retinopathy
Skin: photosensitivity, hyperpigmentation, telangiectasia, erythema
premature signs of aging and progressive neurologic deterioration |
|
|
What are the genetics and cellular characteristics of Trichothiodystrophy? |
autosomal recessive deficiency in proteins involved in nucleotide excision repair, which results in accumulation of UV light induced DNA damage
skin cancer is usually NOT associated |
|
|
What are the clinical features of Trichothiodystrophy? |
PISSS
Photosensitivity Intellectual disability Short stature Sulfur rich brittle hair and nails Scaly fish-like skin (ichthyosis)
|
|
|
What are the genetics and cellular characteristics found in Ataxia-Telangiectasia? |
autosomal recessive mutation in ATM gene (AT mutated) located on chromosome 11
AT mutation has demonstrated that the protein affected is required for maintenance of genome stability
patients have higher frequencies of chromosome and chromatid breaks and rearrangements (disproportionally affecting chromosome 7 and 14); which is responsible for T cell receptor and immunoglobulin regulation (i.e. mantle cell lymphoma) |
|
|
What are the clinical features of Ataxia-Telangiectasia? |
characterized by progressive neurologic dysfunction, cerebellar ataxia, sino-pulmonary infections, telangiectasias of skin and eyes; increased risk of malignancy and hypersensitivity to X-rays
characteristics dull relaxed faces and oculomotor signs
30% have mild mental retardation
skin and hair have accelerated signs of aging |
|
|
Treatment and prognosis of Ataxia-Telangiectasia: |
treatment aimed at controlling infections and malignancies
supportive neurologic care
most patients survive until early or mid-adolescence, usually cause of death being bronchopulmonary infections and cancer |
|
|
What are the genetics and cellular characteristics of Hereditary Nonpolyposis Colorectal Cancer? |
autosomal dominant Genes: MSH2, MLH1 and PMS2 ras mutations can be detected in stool
genes are involved in mismatch DNA repair leads to micro-satellite instability |
|
|
What are the clinical features of Hereditary Nonpolyposis Colorectal Cancer? |
also known as Lynch syndrome patients affected by this disorder have an 80% lifetime likelihood of developing colorectal cancer |
|
|
What is micro-satellite instability? |
change in number of repeating units of micro-satellites in germ line alleles
micro-satellites are stretches of DNA made of short repeating motifs (usually between one and five bases in length) |
|
|
What are the genetics and cellular characteristics of Hereditary Breast Cancer? |
autosomal dominant
mutations involve DNA repair machinery higher frequency of p53 mutations seen
Genes: BRCA1 and BRCA2 these mutations also increase risk of breast tissue cancer in men Ashkenazi Jews have increased frequency of mutations
|
|
|
The malignancies of hereditary breast cancer disproportionately include ___________________. |
Serous adenocarcinomas |
|
|
In regard to hereditary breast cancer, patients with BRCA2 mutations also have significantly higher risk of which types of cancer? |
ovarian, prostate and pancreatic |
|
|
Genetic defects in the enzyme helicase result in what types of disorders? |
Bloom Syndrome Werner Syndrome
|
|
|
What are the genetic and cellular characteristics of Bloom syndrome? |
autosomal recessive
Gene: BLM on chromosome 15 more common in Ashkenazi Jews
mutation causes defect in helicase activity
higher frequency of sister chromatid exchanges and chromosomal (genomic) instability is seen and thought to be caused by overproduction of superoxide radicals |
|
|
What are the clinical features of Bloom syndrome (aka. congenital telangiectatic erythema)? |
growth delay (usually of prenatal onset)
increased risk of malignancy (300x fold)
recurrent respiratory and GI infections due to compromised immunity
telangiectatic erythema often seen in a butterfly facial distribution |
|
|
What are the genetics and cellular characteristics of Werner Syndrome? |
autosomal recessive
Gene: WS (aka. WRN) defect in helicase (involved in DNA repair and general transcription and replication) WRN helicase activity is important not only for DNA repair and recombination, but also for maintaining telomere length and stability
more than 80% of reported cases found in Japan
defect particularly affects connective tissues
|
|
|
What are the clinical features of Werner Syndrome? |
aka. progeria of the adult
characterized by accelerated aging, usually in late teen years
disproportionately aged, including thin, tight, scleroderma-like skin; beaked noses; muscle atrophy; wrinkling; hyperkeratosis; gray hair, and nail dystrophy.
Sources of morbidity and mortality: cataracts, osteoporosis, neoplasias, diabetes mellitus and arteriosclerosis |
|
|
Type of Point mutation: Define Tautomerism. |
modification of a base caused by migration of a proton or hydrogen bond, which results in switching of an adjacent single and a double bond |
|
|
Type of Point mutation: Define Depurination. |
caused by a spontaneous hydrolysis of a purine base (A or G) in such a way that its deoxyribose-phosphae backbone remains intact (creates apurinic site) |
|
|
Type of Point mutation: Define Deamination: |
spontaneous reaction in conversion: C into U (only one that CAN be corrected because it can be recognized by proofreading mechanisms) 5-methyl cytosine into T A to hypoxanthine |
|
|
What are splice site mutations? What is a clinical correlation to this form of mutation? |
insertion of nucleotide bases in certain regions of a gene can result in altering the splicing of introns from the precursor mRNA. The mRNA will contain introns.
Beta-globulin gene -- Beta thalassemia
|
|
|
What is amplification?
What is a clinical pharmacologic correlation to this form of mutation? |
results in multiple copies of whole DNA segments, including all the genes located on them
usually caused by a disproportionately high level of DNA replication in a limited portion of the genome = higher number of copies of encoded protein
drug resistance in certain cancers is linked to amplification of genes that confer resistance to chemotherapeutic agents by preventing their uptake into the cell |
|
|
Define reciprocal (non-robertsonian) translocation. |
True exchange of DNA fragments between two nonhomologous chromosomes. This can lead to formation of new fusion genes, or changed level of expression of existing genes. Can be balanced or unbalanced. |
|
|
Define robertsonian translocation. |
a large fragment of chromosome attaches to another chromosome, but no DNA is attached or returned common robertsonian translocation are confined to acrocentric chromosomes
|
|
|
Define an acrocentric chromosome. |
centromere is located very near to one end of the chromosome usually short arms of these chromosomes contain no essential genetic material ex: 13, 14, 15, 21, and 22 |
|
|
A very small percentage of Down syndrome cases are caused by which type of Chromosomal Translocation? What does this entail? |
Robertsonian translocation
chromosome translocation of long arm of 21 to 14. (about 5% of down syndrome cases) |
|
|
Contrast primary gout and secondary gout. |
Primary gout: hyperuricemia without evident cause may occur due to PRPP synthetase hyperactivity or HGPRT deficiency with Lesch-Nyhan syndrome
Secondary gout: uric acid overproduction--leukemia, myeloproliferative syndrome, multiple meyloma, hemolysis, neoplasma, psoriasis, alcoholism uric acid under excretion -- kidney disease; drugs (aspirin, diuretics and alcohol)
|
|
|
Clinical presentation of Gout. |
monoarticular arthritis of distal joints (i.e. podagra--gout of great toe)
often with hx of hyperuricemia (most hyperuricemia is asymptomatic) precipitated by sudden change in urate levels (due to large meals, alcohol) which eventual leads to nodular tophi (deposits of urate crystals surrounded by fibrous connective tissue) located around joints and achilles tendon. |
|
|
Diagnosis of Gout. |
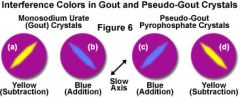
arthritis hyperuricemia detection of negatively bifringent crystals from articular tap negative bifringent crystals = yeLLow when paraLLel to polarized light, blue when perpendicular.
Note: positively birefringent crystals are characteristically found in pseudogout.
|
|
|
Treatment of Gout: |
normalize uric acid levels (allopurinol, probenecid for chronic gout)
decrease pain and inflammation -cochicines and non steroidal anti-inflammatory drugs for acute gout)
avoid large meals, foods rich in purines, red meat and alcohol |
|
|
Histologic imaging of gout: |
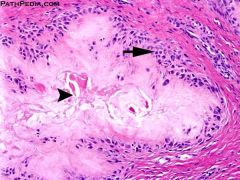
* aggregates of crystalline material
* occasional histiocytes |
|
|
What is often used as an indicator of DNA turnover (increase in chemotherapy or radiation therapy)? |
3-Aminoisobutyric acid (aka. B-aminoisobutyrate) -- product formed by catabolism of thymine |
|
|
Discuss the direction of synthesis of DNA/RNA/protein. |
DNA and RNA are both synthesized 5' to 3' mRNA is read 5' to 3' Protein synthesis is N-terminus to C-terminus.
The 5' end of incoming nucleotide bears the triphosphate (energy source for bond). The triphosphate bond is the target of 3' hydroxyl attack. Drugs blocking DNA replication often have modified 3' OH, preventing addition of the next nucleotide (chain termination). |
|
|
What are the mRNA start codons for prokaryotes and eukaryotes. What do they code for? |
AUG (rarely GUG)
eukaryotes: methionine (may be removed before translation is complete)
prokaryotes: formylmethionine (f-met) |
|
|
What are the stop codons? |
UGA = U Go Away UAA = U Are Away UAG = U Are Gone |
|
|
What is the functional organization of a eukaryotic gene? |
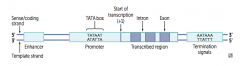
|
|
|
Regulator of Gene Expression: Describe the promoter.
What commonly results from promoter mutations? |
site where RNA polymerase and multiple other transcription factors bind to DNA upstream from gene locus (AT rich upstream sequence with TATA and CAAT boxes)
mutations = dramatic decrease in level of gene transcription |
|
|
Regulator of Gene Expression: Describe the enhancer.
Describe the silencer. |
Enhancer: Stretch of DNA that alters gene expression by binding transcription factors.
Silencer: Site where negative regulators (repressors) bind.
Enhancers and silencers may be located close to, far from, or even within (in an intron) the gene whose expression it regulates |
|
|
RNA polymerase--Eukaryotes: Describe RNA polymerase I, II and, III. |
RNA polymerase I: makes rRNA (most numerous RNA, rampant)
RNA polymerase II: makes mRNA (largest RNA, massive)
RNA polymerase III: makes tRNA (smallest RNA, tiny) |
|
|
Which RNA polymerase opens DNA at promoter site? |
RNA polymerase II
Note: RNA polymerase II cannot initiate transcription by itself, requires transcription factors |
|
|
What substance has the capacity to inhibit RNA polymerase II? |
alpha-amanitin -- found in amanita phalloides (death cap mushrooms) causes severe hepatotoxicity if ingested |
|
|
Describe RNA polymerase--Prokaryotes:
|
1 RNA polymerase (multi-subunit complex) makes all 3 kinds of RNA |
|
|
During eukaryotic RNA processing: What is the initial transcript called and what is it modified into? |
initial transcript is called heterogeneous nuclear RNA (hnRNA) and is then modified to become mRNA.
mRNA is transported out of nucleus to cytosol to be translated |
|
|
During eukaryotic RNA processing: What processes occur in the nucleus following transcription? |
capping of 5' end (addition of 7-methylguanosine cap) -prevents mRNA degradation; allows translation to begin
polyadenylation of 3' end (about 200 A's) -- Poly-A polymerase does not require a template polyadenylation signal = AAUAAA -stabilizes mRNA and facilitates exit from nucleus
splicing out of introns
Capped, tailed and spliced transcript = mRNA |
|
|
Describe mRNA quality control. |
occurs at cytoplasmic P-bodies which contains exonucleases, decapping enzymes and, microRNAs
Note: mRNAs may be stored here for future translation |
|
|
What is mitochondrial RNA polymerase?
What pharmacologic agent inhibits it? |
transcribes RNA from mitochondrial genes
more closely resembles bacterial RNA polymerase than eukaryotic RNA polymerase
inhibited by rifampin |
|
|
What are the steps of Splicing pre-mRNA? |
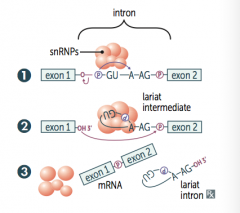
1. primary transcript combines with small nuclear ribonucleoproteins (snRNPs) and other proteins to form spliceosome 2. Lariant-shaped (looped) intermediate is generated 3. Lariant is released to precisely remove intron and join 2 exons
|
|
|
Antibodies to splicesomal snRNPs (aka. anti-smith antibodies) are highly specific for _________.
|
systemic lupus erthematosus |
|
|
Anti-U1 ribonucleoprotein (RNP) antibodies are highly associated with _____________________. |
mixed connective tissue disease
U1 RNP is a snRNP --part of the splicesome |
|
|
Compare introns vs. exons. |
exons contain actual genetic information coding for protein
introns are intervening noncoding segments of DNA
Introns are intervening sequences and stay in the nucleus whereas exons exit and are expressed. |
|
|
Different exons are frequently combined by _______________ to produce a larger number of different unique proteins. |
alternative splicing
NOTE: abnormal splicing variants are implicated in oncogenesis and many genetic disorders (e.g. Beta-globulin mRNA is responsible for some cases of Beta thalassemia) |
|
|
Describe the overall structure of tRNA. |
75-90 nucleotides secondary structure cloverleaf form
|
|
|
Describe the 3' end of tRNA. |
Both eukaryotes and prokaryotes have CCA at 3' end along with a high percentage of chemically modified bases. The amino acid is covalently bound to the 3' end of the tRNA CCA = Can Carry Amino Acids
Acceptor stem: the 3' CCA is the amino acid acceptor site |
|
|
Describe the T-arm and D-arm of t-RNA. |
T-arm: contains the TΨC (thymine, pseudouridine, cytosine) sequence necessary for tRNA- ribosome binding. D-arm: contains dihydrouracil residues necessary for tRNA recognition by the correct aminoacyl- tRNA synthetase. |
|
|
Describe the anticodon end of tRNA. |
opposite the 3' amino-acyl end and is antiparallel and complementary to the codon in mRNA |
|
|
Describe tRNA Charging. |
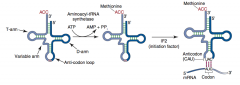
Aminoacyl-tRNA synthetase (1 per amino acid; “matchmaker”; uses ATP) scrutinizes amino acid before and after it binds to tRNA. If incorrect, bond is hydrolyzed. The amino acid-tRNA bond has energy for formation of peptide bond. A mischarged tRNA reads usual codon but inserts wrong amino acid.
Aminoacyl-tRNA synthetase and binding of charged tRNA to the codon are responsible for accuracy of amino acid selection.
Video: https://www.youtube.com/watch?v=0B-CFLNAnX8
|
|
|
Describe tRNA wobble. |
Accurate base pairing is required only in the first 2 nucleotide positions of an mRNA codon, so codons differing in the 3rd “wobble” position may code for the same tRNA/amino acid (as a result of degeneracy of genetic code)
Often, the same amino acid has multiple codons that differ in the third "wobble" position. Therefore, less that 61 tRNAs are actually need to translate all 61 codons. |
|
|
Ribosomes (composed of RNA and protein): Compare Eukaryotic vs. Prokaryotic. |
Eukaryotes: 40S + 60S = 80S (Even)
PrOkaryotes: 30S + 50S = 70S (Odd) |
|
|
Protein synthesis 3 steps: Describe initiation. |
initiated by GTP hydrolysis initiation factors (eukaryotic IFs) help assemble the 40S ribosomal subunit with the initiator tRNA and are released when the mRNA and the ribosomal 60S subunit assemble with the complex
ATP—tRNA Activation (charging). GTP—tRNA Gripping and Going places (translocation). |
|
|
Protein synthesis 3 steps: Describe elongation. |
1. Aminoacyl-tRNA (charged t-RNA) binds to A site (except for initiator methionine) 2. Amino acid in A site forms peptide bond with peptide in P site. Reaction is catalyzed by peptidyl transferase and uses energy from the bond between the amino acid and tRNA. The peptide in P site is effectively transferred to the amino acid-tRNA in the A site, leaving the tRNA in the P site empty. 3. Ribosome advances 3 nucleotides toward 3′ end of mRNA. Uncharged tRNA is now in E site (where it exits) and tRNA which the growing chain enters the P site.
Think of “going APE”: A site = incoming Aminoacyl-tRNA. |
|
|
Protein synthesis 3 steps: Describe termination. |
Stop codon is recognized by release factor, and completed polypeptide is released from ribosome. |
|
|
Concerning bacterial ribosomes: list the drug classes that target specific ribosome subunits. |
Buy AT 30, CELL at 50
Aminoglycosides Tetracycline
Chloramphenicol Erythromycin Linezolid cLindamycin
|
|
|
Posttranslational modifications: Describe Trimming. |
Removal of N- or C-terminal propeptides from zymogen to generate mature protein (e.g., trypsinogen to trypsin). |
|
|
Posttranslational modifications: Describe Covalent alterations. |
Proteins may be covalently modified through: phosphorylation = turns protein on or off glycosylation = proteins that will be secreted or reside in the plasma membrane/lysosomes will be glycosylated hydroxylation = i.e. proline hydroxylation to become component of collagen methylation = i.e. histone methylation acetylation = i.e histone acetylation ubiquitination = targeting for protein degradation |
|
|
What is a chaperon protein? |
Intracellular protein involved in facilitating and/or maintaining protein folding. prevent protein denaturing/misfolding. |