![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
54 Cards in this Set
- Front
- Back
|
Hämhorrhagische Diathese ・Definition (2) |
- angeborene oder erworbene Blutungsstörung - Blutungen treten zu lange, zu stark oder ohne entsprechenden Anlass auf |
|
|
Hämhorrhagische Diathese ・Ätiologie (3) |
-70% Störung der Thrombozyten → Thrombozytopenie → Thrombozytopathie (abnorme Funktion)
- 20% Koagulopathien → Fehlen/Funtionsverlust plasmatischer Gerinnungsfaktoren → va Sugillationen und Einblutungen in Gelenk und Muskel.
- 10% Vaskuläre Genese - Kombinierte Störungen (zb. Verbrauchskoagulopathie) → va petechiale Blutungen |
|
|
Hämhorrhagische Diathese ・Klinik (4) |
- Haut- und Schleimhautblutungen → Petechien (flohstuchartig) → Purpura (fleckig) → Ekychymosen (kleinere bis mittlere Blutungen) → Sugillationen (flächenhafte Blutungen) |
|
|
Hämostase ・Definition (2) |
- aka Blutstillung - Verläuft in zwei Phasen (primäre und 2nd Hämostase) |
|
|
Primäre Hämostase ・Pathophysiologie (7) (Bild) |
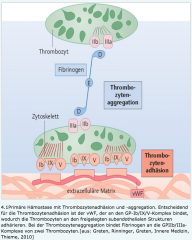
- Verletzung der Gefässwand → Enothelzellen sezerniern Von-Willebrand-Faktor → Bindung an den Glykoprotein(GP)-Ib/IX/V-Komplex der Thrombozyten → Aktivierung und Adhäsion an Endothel → Aggregation (Fibrinogen) und Sekretion → Vasokonstriktion + ↑Aggregation → Instabiler Plättchenthrombus (weisser Thrombus) |
|
|
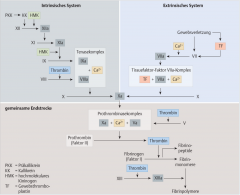
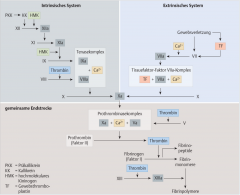
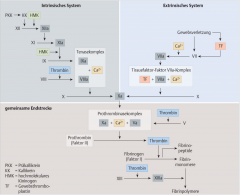
Sekundäre Hämostase ・Pathophysiologie (6) (Bild) |
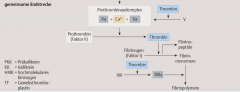
- Aktivierung der extrinsischen/intrinsischen Gerinnungskaskade → Proteolytische Spaltung des Prothrombins zu Thrombin durch den Prothrombinasekomplex → Spaltung von Fibrinogen zu Fibrinmonomeren → Fibrinmonomere polymerisieren und können in Gegenwart von Faktor XIIIa zu stabilen Multimeren verknüpft werden → Erys und Leukos bleiben in diesem Maschwerk stecken → stabiler, roter Abscheidungsthrombus |
|
|
Extrinsische Gerinnungskaskade ・Pathophysiologie (6) (Bild) |
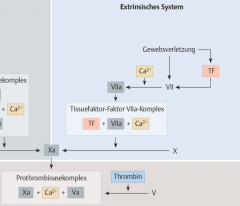
- Gefässverletzung → Freisetzung von Gewebethromboplastin (TF) → Bindung an Faktor VII (Prokonvertin) → Faktor VIIa wird aktiviert (TF-Faktor-VIIa-Komplex) → Faktor X (Stuart-Power-Faktor) wird zu Faktor Xa aktiviert → Bildung des Prothrombinasekomplexes (FXa, FVa, Ca²⁺ und Phospholipide)
|
|
|
Intrinsische Gerinnungskaskade ・Pathophysiologie (4) (Bild) |
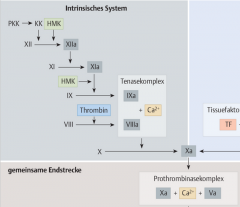
- Aktivierung von Faktor XIIa durch Kalikrein und hochmoleklares Kininogen → Faktor XI (Rosenthal-F) wird aktiviert zu Faktor XIa → Tenaskomplex → Faktor X Aktivierung |
|
|
Gerinnungsfaktoren ・Synthese (2) |
- werden in der Leber synthetisiert → Vit K unabhängig: I, V, VIII, XI, XII, XIII, Präkalikrein, Kininogen → Vit K abhängig: II, VII, IX, X (1927) (Cumarine)
|
|
|
Inhinitoren der Gerinnung ・Übersicht (3) |
- Antithrombin III (AT III) - Heparin - Protein C und Protein S |
|
|
Antithrombin III (AT III) ・Definition (2) (Bild) |
- In der Leber gebildete Protease - Inaktivierung vorwiegend von FIIa (Thrombin) und FXa |
|
|
Heparin ・Einteilung (3) |
- Unfraktioniertes (hochmolekulares) Heparin (UFH) - Niedermolekulare Heparin (NMH) - Fondaparinux |
|
|
Unfraktioniertes (hochmolekulares) Heparin (UFH) ・Definition (5) |
- Standard Heparin - Bildet Komplex mit ATIII und verstärkt antikoagulatorische Wirkung (va Hemmung FXa) - AT-III-Heparin-Komplex kann ausserdem Thrombin inaktivieren - Nicht Plazenta o. Milch-gängig - Antagonisierung mit Protamin |
|
|
Niedermolekulares Heparin (NMH) ・Definition (3) |
- kurzkettiger als UFH (daraus gewonnen) → können keinen Komplex mit AT III bilden → Selektive FXa Hemmung - längere konstante Wirksamkeit - Antagonisierung mit Protamin nicht vollständig möglich
|
|
|
Fondaparinux ・Definition (2) |
- synthetisches Heparin Analogon - Nur Selektive FXa Hemmung |
|
|
Heparin-induzierte Thrombozytopenie (HIT) ・Definition (2) (Tabelle) |
- Häufigste medikamentenassozierte Thrombozytopenie - Eingeteilt in 2 Typen (HIT Typ 1 und HIT Typ 2) |
|
|
HIT Typ 1 ・Definition (4) |
- nicht immunologische Frühform der HIT 2 in den ersten 2d der Behandlung - Klinisch harmlos - Ausgelöst durch proaggregatorische wirkung des Heprains durch Hemmung der Adenylatzyklase - Bei normalisierung kann Heparinisierung normal weitergeführt werden |
|
|
HIT Typ 2 ・Definition (4) |
- einhergehend mit lebensbedrohlichen Thrombosen - Ausgelöst durch AKbildung gegen Heparin-PF4-Komplexe und Plättchenktivierung - Bei HIT 2 Verdacht sofortige Beendigung der Heparinisierung auch ohne AKnachweis - Weiter Antikoagulation (Argatroban o. Danaparoid) durchführen |
|
|
Protein C und Protein S ・Definition (3) (Bild) |
- Vitamin-K abhängig in der Leber synthetisiert - Protein S aktiviert Protein C - Protein C inaktiviert FVa und FVIIIa |
|
|
Störungen der Blutgerinnung ・Rumpel-Leede-Test (3) |
- petechiale Blutungen am Unterarm nach Aufblasen einer BDmanschette auf 90mmHg - positiv bei Thromboztenfunktionsstörungen bzw. ↑Kapillarfragilität - negativ bei Koagulopathien
|
|
|
Thrombozytär bedingte Gerinnungsstörungen ・Überblick (4) |
- Thrombozytopenie - Thrombotische Mikroangiopathien - Idiopathische thrombozytopenische Purpura (ITP) - Thrombozytopathien |
|
|
Thrombozytopenie ・Definition (2) |
- Absinken der Thrombozytenzahl < 140000 Zellen/ul - Bedrohliche Blutungen ab <10000/ul |
|
|
Thrombozytopenie ・Therapie (2) |
- Kausale Therapie je nach Grunderkrankung (bsp Danaparoid bei HIT) - Bluttplättchen Substitution (Indikation <10000/ul) (1 Transfusion + 30000/ul) |
|
|
Thrombotische Mikroangiopathien ・Definition (2) |
- Hämolytisch-urämisches Synrom (HUS) - Thrombotisch thrombozytopenische Purpura |
|
|
Idiopathische thrombozytopenische Purpura (ITP) ・Definition (3) |
- aka M. Werlhof - AutoAK bedingte Thrombozytopenie (< 30000/ul) - Reduzierte Überlebenszeit der Thrombozyten (AK markierte Thrombos werden frühzeitig in der Milz abgebaut (dezente Hepatosplenomegalie) |
|
|
Idiopathische thrombozytopenische Purpura (ITP) ・Einteilung (2) |
- Akute ITP → < 6 Mo → Hauptsächlich Kinder nach GI oder respiratorischem Virusbefall → hoche Spontanremission
- Chronische ITP → > 6 Mo → Geht aus einer akuten ITP hervor → ♀>♂ |
|
|
Idiopathische thrombozytopenische Purpura (ITP) ・Therapie (4) |
- Kortikosteriode 2MG/Kg/d für 14d - Immunglobuline bei hohem Blutungsrisiko - Eltrombopag (↑Thrombozytenproduktion) - Immunsuppressiva (Azathioprin und Vincristin) |
|
|
Thrombozytopathien ・Definition (3) |
- Störung der Thrombozytenfunrktion - oft erworden - selten angeboren |
|
|
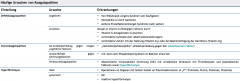
Koaguloathien ・Definition (1) |
- Synthese oder Funktionsstörung eines oder mehrerer Faktoren der Gerinnungskaskade |
|
|
Koaguloathien ・Ursachen (Tabelle) |
|
|
|
Hämophilie ・Definition (1) (Tabelle) |
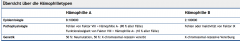
- X-chromosomal-rezessiv vererbter Aktivitätsmangel von FVII (Hämophilie A) und FIX (Hämophilie B) |
|
|
Hämophilie ・Pathogenese (2) |
- Durch FVIII bzw FIX-Mangel Störung der intrisischen Gerinnung → Mangel an aktivem Thrombin → verzögert einsetzende Fibrinbildung - FVIII zusätzlich an Kollagenbildung beteiligt → Hamophilie A Wundheilungsstörungen |
|
|
Hämophilie ・Klinik (2) |
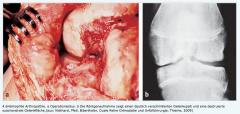
- Grossflächige Blutungen bereits nach Bagatellverletzungen - Hämophile Arthropathie (rezidivierende Hämarthros führen zu destruktiven Veränderungen) |
|
|
Hämophilie ・Schweregradeinteilung (Tabelle) (4) |

|
|
|
Hämophilie ・Diagnostik (3) |
- Verlängerte aPTT (intrinsisches System) - Normale Quick, Thrombin, Blutungszeit - Faktorenaktivität Bestimmung |
|
|
Hämophilie ・Therapie (2) |
- Faktorenkonzentrat Substitution (bei ausgeprägter Symptomatik 20-30IE/Kg) (1IE=1%↑) → Es kann zur AK (IgG gegen FVIII) Bildung kommen (Hemmkörperhämophilie) va postpartal oder bei Autoimmunerkrankungen. (Behandlung mit Prothrombinkomplexkonzentraten) - Desmopressin (DDAVP) → Freisetzung von im Endothel gespeichertem FVIII und vWF (Nur bei leichter Häm A) |
|
|
Von-Willebrand-Jürgens-Syndrom (vWS) ・Definition (2) |
- meist hereditäre Funktionsminderung des vom-Willebrand-Faktors mit eine kombinierten plasmatischen und thrombozytären Gerinnungsstörung - häufigste angeborene Gerinnungsstörung |
|
|
Von-Willebrand-Jürgens-Syndrom (vWS) ・Pathophysiologie (2) |
- 1. Störung der primären Hämostase (Thrombozytenadhäsion) - 2. Störung der plasmatischen Gerinnung (mangelnde Stabillisierung des FVIII-Komlexes) |
|
|
Von-Willebrand-Jürgens-Syndrom (vWS) ・Einteilung (3) (Tabelle) |

|
|
|
Von-Willebrand-Jürgens-Syndrom (vWS) ・Diagnostik (3) |
- Diskrete Blutungen - Verlängerte Blutungszeit (DD: Hämophilie) - Verlängerte aPPT (bei FVIII-Mangel) |
|
|
Von-Willebrand-Jürgens-Syndrom (vWS) ・Therapie (1) |
- Desmopressin (Cave Typ 2B KI, Typ 3 wirkungslos) |
|
|
Disseminierte intravasale Geinnung (DIC) ・AKA (1) ・Definition (2) |
・AKA - Verbrauchskoagulopathie
・Definition - Überaktivierung des plasmatischen Gerinnungssystems - Bildung von Mikrothromben in der terminalen Strohmbahn → Verbrauch von Thrombozyten und Gerinnungsfaktoren (2nd Mangel) → hämmorrhagische Diathese
|
|
|
Disseminierte intravasale Geinnung (DIC) ・Ätiologie (Tabelle) |
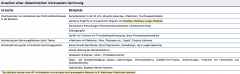
|
|
|
Disseminierte intravasale Geinnung (DIC) ・Pathogenese (4) (Bild) |
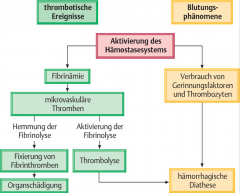
- Aktivierung des Gerinnungssystems → unkontrolierte Freisetzung von plasmatischen Gerinnungsfaktoren und Inhibitoren → multiple Thrombenbildung und ↑Blutungsneigung → ↑Fibrinolyse ("reaktive Hyperfibrinolyse") |
|
|
Disseminierte intravasale Geinnung (DIC) ・Klink (3) |
- Verlauf in 3 Stadien → 1. keine Symptome. Aktiviertes und kompensiertes Gerinnungssytem → 2. frühe Verbrauchsphase. Zuhnemende Blutungen und Organfunktionsstörungen → 3. spähte Verbrauchsphase und reaktive Fibrinlolyse. Ausgeprägte Blutungen und Organfunktionsstörungen
|
|
|
Disseminierte intravasale Geinnung (DIC) ・Diagnostik (7) (Tabelle) |
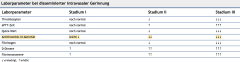
- Thrombozytopenie - verlängerte aPTT - ↓Quick - ↓AT III-Aktivität - ↑D-Dimere - ↓Fibrinogen (erhöt bei SS und Erkrankung) - Nachweis von Fibrinmonomeren (DD: primäre Fibrinolyse) |
|
|
Disseminierte intravasale Geinnung (DIC) ・Therapie (6) |
- Stadium I → niedrig dosiertes Heparin - Stadium II und III → FFP → Antithrombin → PPSB → Thrombozytenkonzentrate (Thrombos < 20000/ul) → Tranexamsäure (Nachgewiesene Fibrinolyse) |
|
|
Thrombophilie ・Definition (1) |
- angeborene oder erworbene Erkrankungen, die mit einem erhöten Thromboserisiko einhergehen |
|
|
Thrombophilie ・Ätiologie (4) (Tabelle) |
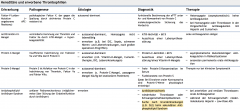
- 30-40% Faktor-V-Leiden-Mutation (APC-Resistenz) - 5% AT III-Mangel - 5% Protein C-Mangel - 5% Protein S-Mangel |
|
|
Faktor-V-Leiden-Mutation (APC-Resistenz) ・Definition (2) |
- Autosomal-dominante Mutation des FV - Pathologischer FV ist resistent gegen die Spaltung durch das Protein C
|
|
|
Faktor-V-Leiden-Mutation (APC-Resistenz) ・Diagnostik (2) |
- aPTT Bestimmung unter An und Abwesenheit des Protein C - molekulargenetischer Nachweis |
|
|
Faktor-V-Leiden-Mutation (APC-Resistenz) ・Therapie (2) |
- Heterozygotie (asymptomatisch): keine Antikoagulation - Homozygotie oder Thrombose in der Vorgeschichte: Antikogulation mit Heparin und Cumarinen |
|
|
Thrombophilie ・Diagnostik (5) |
- Quickwert - aPTT - AT III Aktivität - Purpura fulminans beim Neugeborenen (Hauteinblutungen mit Nekrotischen Veränderungen) - Antiphospholipid-AK (5-15% normal Bevölkerung vs 30% Thrombophilie Pat) |
|
|
Thrombophilie ・Therapie (2) |
- initial Heparin - orale Antikoagulation als Dauertherapie |