![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
399 Cards in this Set
- Front
- Back
|
Définition de nécrose |
-Dénaturation des protéines intracellulaires -digestion enzymatique de la cellule endommagée de façon irréversible. -suite à un stimulus exogène -perte de l’intégrité membranaire, une digestion enzymatique des cellules, un écoulement des composantes cellulaires et une réaction de l’hôte. |
|
|
Différence entre nécrose et apoptose par rapport:
-chromatine: -organites: -distribution: -inflammation?:
|
a- Chromatine : condensation périmembranaire dans l'apoptose et simple pycnose diffuse dans la nécrose suivie de caryorexie.
b- Les organites paraissent en bon état dans l'apoptose alors qu'ils sont lésés dans la nécrose.
c- L'apoptose touche des cellules isolées et dispersées dans un tissu tandis que la nécrose implique généralement des groupes contigus de cellules soumises aux mêmes conditions délétères.
d- L'apoptose ne peut déclencher de réponse inflammatoire alors que la nécrose va généralement la provoquer. |
|
|
Caryolyse |
Diminution de la basophilie de la chromatine lors de la nécrose, reflétant l'activité de la Dnase. (perte de matériel génétique) |
|
|
Pycnose : |
Condensation de l'ADN dans une cellule nécrotique en une petite masse solide |
|
|
Caryorexie : |
Lorsqu'un noyau partiellement ou totalement en pycnose se fragmente. |
|
|
Autolyse : |
Lorsque la nécrose (digestion enzymatique) résulte des enzymes des lysosomes des cellules mêmes. |
|
|
Hétérolyse :
|
Lorsque la nécrose (digestion enzymatique) résulte des enzymes provenant des lysosomes de leucocytes immigrants. |
|
|
5 étapes des lésions cellulaires: |
1) Homéostasie et adaptation 2) Atteinte réversible 3) Réponse fonctionnelle et structurelle 4)Atteinte irréversible (point de non retour) 5) Nécrose et apoptose (selon manifestations morphologiques) |
|
|
4 systèmes intracellulaires sont particulièrement vulnérables aux lésions cellulaires : |
Intégrité des membranes cellulaires Respiration aérobie Synthèse de protéine Préservation du génome |
|
|
Définir les 2 manifestations morphologiques des lésions cellulaires visibles au microscope:
Cellular swelling:
Fatty change: |
Cellular swelling: (enflure, gonflage) -Cellular swelling is the result of failure of energy-dependent ion pumps
Fatty change: -occurs in hypoxic injury and various forms of toxic or metabolic injury. It is manifested by the appearance of lipid vacuoles in the cytoplasm. |
|
|
Facteurs influençant les lésions cellulaires |
- Nature de l'agent - Dose - Durée - Intensité du dommage tissulaire - Susceptibilité de la cellule |
|
|
Atteintes possibles d'une cellule avec une lésion réversible |
-Altérations membrane plasmique (bulle, microvilli) -Mitochondrial changes (swelling) -Dilatation du R.E (détachement polysomes) -Nuclear alterations (éléments fibreux, granulaires) |
|
|
Facteurs provoquant le phénomène de nécrose si touchés |
- Calcium intracellulaire en entrée massive - Défectuosités de la perméabilité de la membrane - Dommages irréversibles aux mitochondries et perte du cytochrome oxydase. Déplétion en ATP. Oxygène et ses dérivés radicaux libres - Protein synthesis. |
|
|
Caractéristiques d'une cellule nécrotique |
* Changements nucléaires (pycnose, caryorexie,caryolyse) * Myelin figures *Lyse cellulaire, membranes leakage |
|
|
Types de nécrose: (6) |
1) Coagulatrice : par hypoxie, fragmentation/ phagocytose (conservation architecture tissulaire) 2)Liquéfiée: Brain, et pus (classique bactérienne/fongique) 3) Gangrène*: combinaison coagulatrice par ischémie M.Inf. (si + infection=wet) 4) Caséeuse: dénaturation + digestion= architecture détruite (tuberculose) Granuleux 5) Adipeuse*: saponification, lipases pancréatiques. Pancréatite 6) Fibrinoid: Complexe immuns, fibrine, vaisseaux, éosino. |
|
|
V-F Hypoxie (pas d'O2) est plus dommageable que l'ischémie (blocage sanguin). |
Faux: -Contrairement à l'hypoxie, durant l'ischémie, la production d'énergie via le glucose (glycolyse) ne peut pas continuer : elle s'arrêtera aussitôt que les substrats glycolyliques seront épuisés, ou lorsque l'accumulation de métabolites (qui seraient normalement évacués avec le débit sanguin) est trop grande. Pour cette raison, l'ischémie est plus dommageable que l'hypoxie. |
|
|
Cascade hypoxique |
-RElisse + Mitochondrie= - Haut calcium intracellulaire + pas d'o2= - dommage de la membrane mitochondriale = - pore de transition de perméabilité mitochondriale= - perte du potentiel mitochondrial: - Déplétion en ATP
- Haut calcium = activation enzymes
-Enzymes, ROS = Membrane damage
-ROS = Protein misfolding
|
|
|
Comment juger si l'atteinte hypoxique est irréversible: |
Se caractérise par
-Incapacité de contrer les dysfonctionnements mitochondriaux suite à un trop grand manque d'ATP -Développement de profondes altérations au fonctionnement membranaire. |
|
|
V-F La reperfusion tissulaire suite à une ischémie signifie l'arrêt de processus dommageable pour les tissus. |
Faux, -réaction autocatalytique des radicaux libres Les radicaux libres créés et accumulés suite au manque d'O2 sortent de la cellule, entrent dans la circulation, vont dans les tissus voisins et créent des lésions dans les tissus voisins. |
|
|
Intoxication au tétrachlorure de carbone. |
-Activation dans le REL -le CCl3 peroxyde les lipides -entraîne une stéatose hépatique suite à une baisse d'apoprotéines -puis entrée massive de Ca |
|
|
Intoxication aux tylénols |
-Oxydation en métabolite électrophile, -réduction des niveaux de GSH (glutathion), -nécrose hépatocellulaire massive, -peroxydation des lipides (contrée par les antioxydants). |
|
|
Sources et effets des radicaux libres: |
sources: -radiations, Réaction inflammatoire (NAPDH-oxydase), Métabolisme de drogues, Processus métaboliques normaux (oxydo-réduction), Métaux de transition, NO (oxyde nitrique)
effets: -Peroxydation des lipides membranaires -Oxydation de protéines : fragmentation, dénaturation, dégradation -Lésions de l'ADN |
|
|
Mécanismes de défense vs. Les radicaux libres |
-Antioxydants -Protéines de transport (ferritine, transferrine, lactoferrine, céruloplasmine) et baisser niveau Fer et Cuivre. -Enzymes (catalase, superoxyde dysmutase) |
|
|
Pourquoi n'y-a-t-il pas d'inflammation dans l'apoptose? |
Contenu cellulaire ne coule pas dans le liquide interstitiel par l'intégrité de la membrane plasmique.
*Avantage compte tenu de sa survenu dans des situations physiologiques naturelles. |
|
|
Dans quelles situations pathologiques, l'apoptose occure-t-elle? |
-Dommages d'ADN: Radiations, médicaments, drogues, hypoxie
- Accumulations de protéines ayant une mauvaise configuration 3D (Entraine un Stress du réticulum endoplasmique qui entraine l’apoptose
-Mort cellulaire dans certaines infections (Surtout virales [morts peuvent être causées par le virus lui-même, ou par la réponse de l’hôte] )
-Atrophie des organes du parenchyme après une obstruction des canaux (Pancréas, glande parotide...) |
|
|
Caratéristique évidente de l'apoptose d'un point de vue morphologique |
Condensation périphérique de la chromatine sous la membrane nucléaire.
*noyau peut se diviser+ bourgeonnement et corps apoptotiques. (flipp lipidiques)
-rétrécissement cellulaire |
|
|
Changements biochimiques durant l'apoptose:
|
-Activation des caspases
-(Clivage de l’ADN) et des protéines
-Altération de la membrane et reconnaissances par les phagocytes. Flip lipidique (La membrane se modifie POUR être reconnue) |
|
|
Mécanisme de l'apoptose= Quelles sont les 2 voies possibles? Acteurs en jeu? |
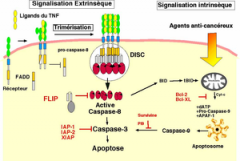
|
|
|
Quelle est l'importance d'éliminer les cellules mortes, par des changements membranaires + phagocytose? |
Éviter une nécrose secondaire. *Processus: -phospo sur feuillet externe - sécrétion de facteurs (thrombospondine) -Apparition d'anticorps et protéines du système complémentaire |
|
|
Quels événements biopathologiques causent l'apoptose? |
Intrinsèque: 1) Privation facteur de croissance: voie intrinsèque (Bcl-2, Bcl-x, Bim et autres) 2) Dommages à l'ADN: p53- Bax,Bak, BH3 3) Mauvaise configuration des protéines: chaperon + ubiquitine ou +/- caspases
Extrinsèque: 4)Récepteurs à TNF 5) Lymphocytes T cytotoxiques (perforine, granzymes, FasL-Fas) 6) Apoptose non-régulée (survie ou mort cellulaire) |
|
|
Hétérophagie |
Des matériaux extérieurs sont détruits suite à une endo/phagocytose transformée en phagolysosome. |
|
|
Autophagie |
Lorsque les matériaux dégradés proviennent directement de la cellule qui a formé un autophagolysosome à l'aide du RER (formé à partir de la membrane libre de ribosomes) fusionné à un lysosome. Sert lorsque les organites sont endommagés, dans le remodelage et différenciation cellulaire ou lorsque l'apport nutritif est insuffisant.
*Peut être un avantage pour les cancers. |
|
|
Nommez quelques enzymes importantes des lysosomes produites par R.E, transportées par Golgi. |
▪ Lipase, qui dégradent les lipides en acides gras. tripeptides, dipeptides, puis acides aminés.
*tout le matériel nécessaire pour dégrader une cellule |
|
|
L'hypertrophie du réticulum endoplasmique lisse signifie quoi? |
l'adaptation à un médicament. - via Cyp-450, (hypertrophie du réticulum lisse) - Mécanisme oxydatif amenant problèmes malgré la détoxication augmentée |
|
|
Dans quelle situation trouvons-nous des mégamitochondries? |
présente chez des foies d'alcooliques et lors de certaines déficiences nutritionnelles.
Mitochondrie de taille et forme anormalement grosse, |
|
|
Que sont les corps de Mallory? |
-Altération des filaments intermédiaires. -Se caractérise par des inclusions éosinophiles intracytoplasmiques de filaments de kératine dans les cellules hépatiques atteintes par l'alcool. |
|
|
Définition de la stéatose: |
Accumulation anormale de triglycérides dans les cellules du parenchyme. Est souvent vue dans le foie et le cœur, mais peut aussi avoir lieu dans les muscles et les reins.
***Synthèse accentuée de lipides, mais moins de dégradation***
-peut être dû à l’alcoolisme ou l’éthylisme -toxines -malnutrition -diabète -obésité -anoxie (et non hypoxie) |
|
|
V-F Certains lipides ne peuvent s'accumuler dans une cellule |
Faux, toutes les classes majeures de lipides peuvent s'accumulées (FFA, 3G, Lipoprot, cholestérol, phospholipides) -Faible: aucun effet sur fct cellulaire -Plus sévère: peut engendrer mort cellulaire, couleur du foie, tigered effect du myocarde |
|
|
Causes possibles d'accumulation de protéines (intra ou extra) |
-Réabsorption de gouttelettes protéique au niveau des cellules du tubule rénal - Problèmes de transport et de sécrétion protéique - Accumulation de protéines qui forment le cytosquelette - Accumulation de protéines anormales |
|
|
Exemple classique de polyoside:
Comment on peut le détecter dans une cellule? |
Glycogène -Traitement à l'amylase. |
|
|
V-F La stéatose, l'accumulation d'eau et l'accumulation de polyoside sont tous les 3 réversibles. |
Faux, l'accumulation de polyoside (glycogène) correspond à une maladie d'accumulation cellulaire par déficience enzymatique progressive et non-réversible |
|
|
Que seraient des granules jaune-bruns autour du noyau présents chez les patients souffrant de malnutrition sévère et de cachexie due au cancer, ou foie et cœur de personnes âgées. |
Pigments de lipofuscine (lipochromes) :
-des polymères de lipides et phospholipides complexés à des protéines et représentent du matériel indigeste (lipides polyinsaturés de membranes subcellulaires) suite à la peroxydation intracellulaire. |
|
|
Expliquez l'évolution des hémorragies, en bleu vers le brun. |
-Les macrophages phagocytent les débris de globules rouges et transforment le fer de l'hémoglobine en hémosidérine (créant un "bleu" qui devient "brun"). Ferritine est à l'origine des granules d'hémosidérine lorsqu'il y a un excès de fer.
-l'HÈME va former=pigment jaune-vert, la bilirubine.
-la globine sera hydrolysée en acides aminés simples par les enzymes lysosomiales.
|
|
|
Où se trouvent habituellement les granules d'hémosidérine?
|
Se retrouve normalement dans les phagocytes mononucléaires de la moelle osseuse, dans la rate et le foie. (résultat de la dégradation des globules rouges) relachement de fer...
Peut évoluer en hémosidérose systémique si se retrouvent dans parenchyme foie, pancréas, coeur |
|
|
2 sortes des dépôts tissulaires de calcium avec fer, magnésium et minéraux. |
1) Calcification dystrophique: local, tissus mourrants, taux normal calcium, pas de prob du métabolisme= zones de nécrose. Athérosclérose
2) Calcification métastatique: dépôts dans tissus normaux (principalement ceux sécrétant acide et compartiment interne alcalin), hypercalcémie secondaire à un problème du métabolisme calcique. |
|
|
Pathogénèse de la formation de cristaux de calcium |
Initié par dommage membranaires: oLiaison du calcium aux phospholipides de la membrane vésiculaire oLes phosphatases de la membrane génèrent du phosphate qui se lie au calcium oCe cycle est répété, ce qui augmente la concentration de calcium et produit un dépôt oFormation de cristaux qui se propage. |
|
|
4 Causes principales d'hypercalcémie: |
1)Augmentation de la sécrétion de la parathormone qui entraine une résorption des os 2)Destruction des os secondairement à une tumeur de la moelle, à une maladie ou à un repos prolongé 3) Problèmes reliés à la vitamine D 4)Insuffisance rénale qui entraine la rétention de phosphate 5) Intoxication aluminium
Aussi : intoxication à l’aluminium |
|
|
Quels sont les organes cibles habituels des calcifications pathologiques survenant chez un patient hypercalcémique? |
*Sécéteurs d'acide* muqueuse gastrique (HCL) Reins- acides alimentaires Poumons- acide carbonique [peut causer des problèmes respiratoires] Artères systémiques et Veines pulmonaires: échange acido-basiques du sang |
|
|
3 facteurs influençant la taille des populations cellulaires. |
1) Vitesse de la prolifération cellulaire: stimulation physiologique ou pathologique. Prolifération par stimulants/inhibiteurs
2) Différenciation cellulaire: définitive si réplication impossible (neurones, cardiomyocytes)
3) Mort cellulaire par apoptose: |
|
|
3 types de cellules selon leur activité proliférative. |
1) Labile tissus ou continuelle: prolifération grande (peau)
2) Stable tissus: basse prolifération (reins, foie, pancréas= endothélium)
3) Permanent tissus: pas de division (neurones) |
|
|
Étapes générales du cycle cellulaire |
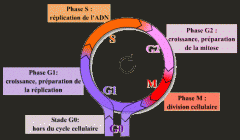
*Importance des facteurs de croissance et des composantes de la matrice extracellulaire via les intégrines. |
|
|
Qu'est-il nécessaire de G1 à S : |
Vérification de l'intégrité de l'ADN, point de restriction important.
CDK4-D phosphoryle RB qui libère E2F |
|
|
Qu'est-il nécessaire de G2 à M: |
Vérifier si l’ADN est endommagée ou non répliquée |
|
|
Étapes de l'activation de la réplication |
oLiaison d'un facteur de croissance à un récepteur (transmembranaire, cytosolique ou nucléaire), comme les tyrosine-kinases, les récepteurs couplés à une protéine G ou les récepteurs sans activité tyrosine-kinase intrinsèque
o Conversion des signaux extracellulaires en signaux intracellulaires:MAP-kinases et IP3; AMP cyclique et JAK/STAT
o Cascade de phosphorylation de protéines (amplification du signal)
oTransfert de l'information au noyau où entrent en jeu des facteurs de transcription qui augmentent ou diminuent la transcription d'un gène |
|
|
Exemple de l'utilité des CDK (cyclin-dependent kinases) à un point de restriction comme celui à G1-S.
que régule l'activité des CDK |
Retinoplasma susceptibility protein; qui prévient la réplication cellulaire. Sa phosphorylation par un complexe cyclines-CDK entraine le relâchement du complexe qu’elle forme pour inactiver E2F, ce qui active E2F et permet la stimulation de la transcription.
*Régulation par inhibiteurs de CDK *Certains facteurs de croissance inhibent la production de ces inhibiteurs. (lol) |
|
|
Principaux facteurs de croissance: bref résumé -EGF -TGF -HB-EGF -HGF -VEGF |
-EGF: Epidermal-Mitogénique (épitheliales, hepatocytes, fibroblastes, migration kératinocytes, tissus granuleux)
-TGF-alpha: Transforming- similaire à EGF, transformation cell. normales en cancer
-HB-EGF: Heparin-Binding: réplication keratinocytes
-HGF: hepatocyte and epithelial growth: Récep dans tumeurs rein et thyroïde.
-VEGF: Vascular endothelial : perméabilité vasculaire, angiogénèse |
|
|
Principaux facteurs de croissance: bref résumé (part 2) -PDGF -FGF -TGF -KGF -TNF |
-PDGF: platelet-derived. Chimiotaxie des PMN Leukocytes. Macrophages, Fibroblastes, MétalloprotéaseMatricielle. Contraction des blessures
-FGF: Fibroblast growth 1(acide)/ 2(basique). Rep des blessures, angiogénèse, hématopoïèse, muscles squelettiques/cardiaques, poumons, spécification du foie à partir endoderme.
-TGF-B: transforming growth: inhibiteur pour epitheliales (MMP et keratinocytes) , fibrinogène (collagène, fibronectine et protéoglycans) Anti-inflammatoire forts, mais peut augmenter certaines défenses!
-KGF: Keratinocytes: migration, prolif et différenciation des kératinocytes
-TNF: tumor necrosis factor: activation macrophages, régulation cytokines, fcts multiples. |
|
|
Rôles et composates de la ECM. (Matrice extra-cellulaire, autant interstielle que la membrane basale) |
- CAMs et Intégrine -Protéines structurales fibreuses (collagène) -Glycoprotéines adhésives -Protéoglycan et hyaluronique ac.
Plusieurs rôles: o Support mécanique de la cellule, migration cellulaire et conservation de la polarité cellulaire o Contrôle de la croissance cellulaire o Conservation de la différenciation cellulaire o Échafaudage pour le renouvellement des tissus o Établissement des micros environnements tissulaires o Storage et préservation de molécules régulatrice (ex : facteurs de croissance) |
|
|
V-F Le collagène type IV, est différent des autres, n'étant pas composé de 3 chaînes formant un hélice et d'un composé d'hydroxyproline stabilisant l'hélice. |
faux, Le type IV, étant à l'origine de la membrane basale, diffère parce qu'il fait des feuillets au lieu de fibres, mais il comporte la même structure de triple hélice que les autres collagènes. (I, II, III, V, XI) *vitamine C nécessaire pour synthèse |
|
|
V-F Le collagène assure la stabilité et l'élasticité des tissus |
faux, non-sens... Ce sont les fibres élastiques qui assurent l'extension et le replacement. (v.sanguins, peau, utérus, poumons. *Élastine au centre, entourée de microfibres (échafaudage) |
|
|
Protéines d'adhésion cellulaire ou CAMs. Liens entre même famille ou différente. Types/utilités:
-Intégrines: -Fibronectines: -Laminines: -Cadhérines: |
-Intégrines: coagulation, prolifération, anti-apoptose, connexion entre cellules et matrice extracellulaire (fibronectine), mais aussi inter-cellules
-Fibronectines: Grosse protéines. Tissulaire / Plasma: fibrine pour caillots et matrice provisoire durant guérison
-Laminines: lame basale, matrice extra et récepteurs surface cellules. réseau serré avec collagène type 4. Alignement cellules et connexion aux substrats.
-Cadhérines: jctn entre cellules même type. zonula adherens (petite, apicale) . desmosomes (fortes, extensives) |
|
|
V-F Toutes les CAMs se lient au cytosquelette (actine et fil. intermédiaires) pour faire signaux de tranduction avec facteurs de croissances |
faux, surtout cadhérines et intégrines, liaison de la surface de la cellule avec cytosquelette: -Transmission de force et messages cellulaires -Complexe d'adhésion focale -Signaux de transduction avec facteur de croissance |
|
|
Quelle est la différence entre glycosaminoglycans et protéoglycans |
Protéoglycans sont des glycosaminoglycans liés à des protéines. -Agissant comme modulateurs dans la réponse inflammatoire, dans la croissance et la différenciation -4 familles : (Heparan Sulfate, Chondroitin/dermatan sulfate, Keratan sulfate, Hyaluronan (HA) ) |
|
|
L'activité des facteurs de croissance et celle des molécules matricielles ont 2 voies intra-cellulaire sur la croissance et la différenciation cellulaire. Lesquelles |
1) Cytoplasmic signal, transduction pathways 2) Cytoskeleton-mediated signals
Dans les 2 cas, le noyau utilise le signal et influence: la prolifération, la différenciation, synthèse protéique, la migration, changement de forme, attachement |
|
|
Hyperplasie |
Augmentation du nombre de cellules dans un organe ou un tissu qui aboutit souvent à l’augmentation de la taille et de la masse de l’organe. Inclut synthèse d’ADN. |
|
|
Hypertrophie |
Augmentation de la taille d’une cellule qui entraine une augmentation de la taille de l’organe concerné. Inclut synthèse d’ADN. Causé par une augmentation de la synthèse de protéines. |
|
|
Atrophie |
Diminution de la taille, du nombre et de l’activité métabolique d’une cellule qui induit une réduction de la taille de l’organe concerné. Causée par la diminution de la synthèse de protéines et aussi par la dégradation de celles-ci. |
|
|
Métaplasie |
Changement réversible dans lequel un type de cellule différenciée est remplacée par un autre type de cellule. C’est un changement dans le phénotype des cellules. Inclut synthèse d’ADN. |
|
|
2 types d'hyperplasie: |
hormonale: -capacité fonctionelle d'un tissu (seins puberté)
compensatoire: -après un dommage ou résection partielle |
|
|
Exemples d'hyperplasie pathologique: |
Excès d'hormones ou facteurs de croissance 1) Endomètre: trop d'oestrogène- saignements excessifs
2) Prostate: trop d'androgènes
|
|
|
V-F Le cancer est une hyperplasie |
faux, cancer a une mutation, donc différent.
toutefois, hyperplasie peut favoriser apparition de cancer.
*hyperplasie fréquente si virus dégage facteurs de croissance |
|
|
V-F L'hypertrophie est la seule manière que les cellules permanentes (neurones, cardio) ont de croître |
vrai. -synthèse de plus de composants structurels - Classique: muscle par la demande, aug du travail - induite par l’action combinée de senseurs mécaniques sur la paroi des cellules, de facteurs de croissance et d’agents vaso actifs |
|
|
Exemple parfait de compensation d'hypertrophie maximale, entrainant des lésions, parfois même nécrose ou apoptose |
Une insuffisance cardiaque qui illustre comment une adaptation au stress peut progresser jusqu'à une lésion cellulaire si le stress n’est pas éliminé. |
|
|
V-F Les organelles peuvent s'hypertrophier |
vrai, exemple du réticulum endoplasmique lors de la prise de médicament. (plus d'enzymes pour détoxification, mais habituation, moins grande réponse) |
|
|
Causes de l'Atrophie |
-Charge de travail diminuée : Bras cassé. Réversible, mais peut emmener une résorption osseuse à long terme -Atrophie de dénervation : Le fonctionnement normal du muscle squelettique dépend de son innervation -Diminution de l'apport sanguin (ischémie) -Malnutrition -Perte de stimulation endocrine : seins, organes reproducteurs dépendent de la stimulation endocrine (ménopause) -Atrophie sénile : le vieillissement est associé avec des pertes cellulaires. -Pression : tumeurs, entraîne ischémie |
|
|
Étapes de l'installation de l'atrophie guidant vers apoptose |
1) diminution de la taille des cell. et organelles, réduisant besoins métaboliques de la cellule 2)pas mortes, mais pas fortes. (destruction protéique par ubiquitine/protéasomes + autophagie lysosomales) 3) si adaptation impossible, progression vers la mort par apoptose 4) remplacement par des cellules adipeuses |
|
|
Exemples de métaplasie: |
1) Prismatique à squameuse: suite à irritation chronique (tabac), vitamine A, pierre au foie, pancréas ou glandes salivaires= épithélium squameux plus résistants, mais sensibles infections. (transformations malignes possible)
2) squameuse à prismatique: en raison de reflux, épithélium intestinale dans l'oesophage. Oesophage de Barret. Risque de carcinomes et adénocarcinomes
3)Métaplasie de tissus: os,cartilage ou gras dans tissus pas supposé. |
|
|
Quels sont les quatre systèmes biochimiques intracellulaires les plus vulnérables à une agression? |
A) Le maintien de l’intégrité des membranes C) La respiration cellulaire aérobique |
|
|
Quels sont les signes morphologiques microscopiques qui permettent d'identifier la nécrose cellulaire? |
Karyorrexia->Pycnosis->Karyolysis |
|
|
- Dans une lésion hypoxique, quels sont les deux événements critiques du seuil d'irréversibilité et quel est le rôle du calcium dans ceux-ci? |
A) Déplétion en ATP -> arrêt de fonction de NaK-ATPases, accumulation de Ca++ dans la cellule. Pas de potentiel mitochondrial
B) Perte de l’intégrité des membranes (attachement du calcium aux phosphates qui composent les membranes). |
|
|
Quels sont les effets biochimiques principaux des radicaux libres sur les cellules? |
A) Peroxidation des lipides C) Lésions sur l’ADN |
|
|
Quels sont les deux processus chimiques en compétition dans la nécrose? |
A) La dégradation enzymatique de la cellule B) La dénaturation protéique |
|
|
Énumérez trois processus physiologiques auxquels l'apoptose contribue. |
A) Embryogénèse C) Destruction des lymphocytes auto-immunes |
|
|
Nommez deux altérations subcellulaires et une forme d'accumulation intracellulaire caractéristiques de l'éthylisme. |
A) -Changement des fonctions mitochondriales et microsomales, qui mènent à une augmentation dans la synthèse et une réduction dans le catabolisme des lipides. -Altération des filaments intermédiaires. (corps de mallory)
B) Stéatose (accumulation des triglycérides) |
|
|
Quels sont les modes d'initiation d'une calcification dystrophique? |
A) Nécrose (coagulante, liquéfactive, caséeuse, graisseuse) |
|
|
Quelle est la différence de distribution du pigment entre la phase initiale et la phase avancée d'une hémosidérose systémique? |
Phase initiale: Hémosidérose se trouve dans les phagocytes mononucléaires dans le foie, rate, moelle osseux, et ganglions lymphatiques, ainsi que dans autres macrophages dispersés dans autre organes
Phase avancée: Hémosidérine se trouve dans les cellules parenchymateuses des organes du corps, surtout le foie, pancréas, et cœur. |
|
|
D'où provient la lipofuscine? |
Lipofuscine provient de la péroxydation des lipides dans les membranes subcellulaires, suggérant une lésion cellulaire par ROS. |
|
|
Dans quels organes la lipofuscine est-elle retrouvée en quantité les plus importantes? |
Au niveau du foie et du cœur |
|
|
Quelle différence y a-t-il entre une cellule stable (quiescente) et une cellule labile ? |
Capacité proliférative: -Labile, c'est continuel -quiescente, en cas d'agression ou de besoin |
|
|
Donnez un exemple de facteur de croissance favorisant la croissance des cellules épithéliales et un autre exemple inhibiteur de croissance. |
Favorise : EGF (tiens donc, ça veut dire EPIDERMAL growth factor)
|
|
|
Décrivez la physiopathologie de l'hyperplasie compensatrice post-hépatectomie |
-Une dégradation de la matrice extracellulaire active les récepteurs de HGF associés à la matrice. -Certaines hormones, comme la norépinephrine, l'insuline, le glucagon et les hormones thyroïdiennes sont des adjuvants dans la prolifération cellulaire. -Le nombre de cellules synthétisant de l'ADN augmente 12 heures après l'hépatectomie, pour atteindre un peak 1 ou 2 jours après où environ 10% des hépatocytes travaillent à la synthèse d'ADN. -La prolifération cellulaire est dépendante des facteurs de croissance et des cytokines. ‐ HGF : hepatocyte growth factor, joue un rôle mitogène avec l'EGF et le TGF-alpha ‐ Cytokines : Interleukine-6 et TNF-alpha
La prolifération est ralentie par les facteurs de croissance inhibiteurs, une baisse des adjuvants et des facteurs de croissance. |
|
|
L'œdème cellulaire peut-il être considéré comme une forme d'hypertrophie ? |
Non, étant donné que l'augmentation de taille des cellules hypertrophiques est due à une synthèse d'un plus grand nombre de structures et non d'une enflure. |
|
|
Lorsqu'une atrophie atteint le point où les cellules meurent, quel changement histologique peut survenir dans ce tissu ? |
Le tissu atrophié mort peut être remplacé par des tissus adipeux. |
|
|
Quel lien y a-t-il entre la lipofuscine et l'atrophie ? |
Les signaux induisant l'atrophie sont aussi ceux induisant l'apoptose. Dans les tissus atrophiés se trouve parfois des vacuoles autophagiques dans lesquelles se trouvent des composantes cellulaires à être digérées par la cellule même. Les parties résiduelles de cette digestion sont les granules de lipofuscine, créant une atrophie brune. |
|
|
2 caratéristiques morphologiques des lésions réversibles |
Swelling + Fatty change |
|
|
Qu'est -ce qui cause la réaction inflammatoire de la nécrose? |
Enzymes lysosomales se déversent dans le cytoplasme et digèrent la cellule et la membrane. Déversement du contenu dans le milieu extra. |
|
|
Qu'est-ce qui est à l'origine des myelin figures? |
Masses phospholipidiques verticillées dérivées de membranes endommagées.
*phagocytées par autres cellules ou dégradées en acides gras (pouvant se calcifier) |
|
|
V-F Ischémie et hypoxie sont synonymes |
Faux, ischémie est en partie une hypoxie fait par obstruction mécanique d'un vaisseau sanguin (artériel ou veineux)
Mais pire: même pas de respiration anaérobique possible. (pas de substrats de la glycolyse) |
|
|
Décrire la lésion d'ischémie/reperfusion |
Tissu irréversiblement endommagé est reperfusé, il y a aura une encore plus grande perte cellulaire: -radicaux libres. oxydative burst. -accumulation par baisse de flot sanguin |
|
|
Réactions d'oxydoréduction lors des processus métaboliques menant à la formation des ROS. |
Transfert d'électrons de l'O2 à des molécules de H2 par NADPH: -Superoxyde ;;;;;SOD le rend en:
*ne pas oublier les diverses causes d'apparition de ROS (énergie, R.inflammatoire, chimique exogènes, métaux, oxyde nitrique) |
|
|
Pathologies cellulaires reliées à l'apparition de ROS: |
- Peroxydation des lipides membranaires (peroxydases lipidiques) - modifications oxydatives des protéines (site actif // protéasomes pour dégradation) -Lésions de l'ADN, mutations
*déclenchement de l'apoptose possible. |
|
|
Dans l'équation de Starling, de quoi dépend le mouvement du sang dans les vaisseaux? |
Pression hydrostatique différentielle. Pression oncotique différentielle. Perméabilité de la membrane capillaire.
1 (sortie) et 2 (entrée) s'oppose. *à l'entrée (artériole), 1 est plus grand que 2: filtration * à la sorti (veinule), 2 plus grand que 1: réabsorption |
|
|
Qu'est-ce qui peut ramener l'excédant de liquide interstiel dans la circulation sanguine? par où? |
Les vaisseaux lymphatiques et retournera éventuellement dans le sang au niveau de la veine sous-clavière gauche via le canal thoracique. |
|
|
Localisation de l'oedème? |
-Hydrothorax (plèvre) -Hydropéritoine (péritoine): Ascite - Anasarca (oedème sévère et généralisé)
|
|
|
Pourquoi la perfusion rénale est très importante dans la formation d'oedème. |
Baisse de la perfusion rénale par défaillance= - RAA system - Retention d'eau - hausse volume sanguin (hausse pression hydrostatique + dilution, baisse pression oncotique) - oedème et défaillance cardiaque accentuée |
|
|
Transudat vs exsudat: |
transudat: pauvre en protéine exsudat: riche en protéines |
|
|
V-F La pression hydrostatique varie selon la position. (couchée vs debout) |
Vrai. bien plus grande quand debout. Comparaison avec niveau de l'oreillette droite. (zéro) |
|
|
Manifestation fréquente du syndrome néphrotique. (perte de l'albumine dans les urines) |
œdème périorbital, caractéristique des œdèmes rénaux. Se manifeste souvent dans les tissus avec un tissu conjonctif lousse comme celui des paupières. |
|
|
V-F Oedème pulmonaire est associé à une insuffisance du coeur droit. |
Faux, coeur gauche, augmente pression dans veines pulmonaires. accumulation de fluides (air,oedème, cellules) dans les septas alvéolaires. |
|
|
V-F Un oedème se manifeste par un gonflement intracellulaire |
faux, gonflement interstitiel et non au niveau des cellules. |
|
|
Différences entre hyperhémie et congestion vasculaire? |
Hyperhémie: hausse afflux sanguin: dilatation artériolaires, erythème (exercice ou inflammation)
Congestion: flot sortant réduit. (systèmique ou local) *cyanose par accumulation d'erythrocytes et mort désoxygénation de l'hémoglobine *possible hémorragies par bris des capillaires (haute pression congestive) |
|
|
Types de congestion: |
1) Congestion pulmonaire aigue: engorgement cap., oedème inter., hémorragie intra
2) Congestion pulmonaire chronique: septum fibreux, macrophages et hémosidérine
3) Congestion hépatique aigue: veine centrolobulaire et sinusoides dilatés. ischémie centro, périportaux ok (artèrioles hépatiques)
4) Congestion passive et chronique: Nutmeg liver (bronzé) non-congestionné en périphérie, espace centrolobulaire en nécrose. |
|
|
Qu'est-ce que l'hémostase? |
L’hémostase est le processus visant le maintien de la fluidité du sang dans un vaisseau sanguin par l’intermédiaire de la formation d’un clou plaquettaire |
|
|
Séquence normale de l'hémostase. (étapes) |
1) Vasoconstriction (réflexe neurogénique + endothéline et Txa2)
2) Adhesion (VwF- Gp1b), Activation (TNF, Il-1) Aggregation (ADP+Fibrinogène-GpIIb-3A)
3) libération du facteur III par cell.endothéliales. activation thrombine (qui active fibrine) 4) Fibrine + plaquettes : hémostase permanente secondaire
*viendra le thrombus et événement antithrombiques (plasmine) |
|
|
Différentes granules et contenus des plaquettes |
- Granules alpha: fibrinogène, fibronectine, facteur V, VIII, (PDGF et TGT-B)
- Granules denses: ADP, ATP, calcium ionisé, histamine, sérotonine, épinéphrine |
|
|
Importance de l'ADP et du calcium dans le phénomène pro-thrombose |
ADP: récepteurs GpIIB-IIIa actif. Liaison au fibrinogène
Calcium activation des facteurs de coagulation ( I, II, V, X, XIII )
*activation des plaquettes= phospholipides négatifs se liant au calcium pour faire la structure du caillot. |
|
|
V-F Le vasoconstricteur TxA2 des plaquettes rend une aggrégation irréversible. |
faux, son agrégation est réversible, c'est toutefois l'Activation simultanée de la thrombine qui rend le tout irréversible |
|
|
Rôles multiples de la thrombine dans l'hemostase une fois liée aux récepteurs activateurs de protéases. (PAR liés à G protéine) |
-Adhésion des neutrophiles (inflammation - aggrégation de fibrine irréversible -activation de l'endothélium (NO, PGI2, tPA) - Aggregation plaquetaire (TxA2) -activation de lymphocyte -activation monocytes (PDGF: muscles lisses) |
|
|
Éléments liés à la cascade intrinsèque de coagulation: |
XII (Hageman), XI, IX, VIII
X, V et II (commun au 2 voies) |
|
|
Éléments liés à la cascade extrinsèque de coagulation: |
III (tissue factor), VII
X, V et II (commun au 2 voies) |
|
|
Triade de Virchow à l'origine de la formation d'un thrombus |
1) Blessure à l’endothélium (moins de PGI2, t-PA)
2) Stagnation ou flot turbulent (facteurs de coagulation stagne)
3)Hypercoagulabilité du sang (innée: facteur V ou acquise:stagnation, contaceptifs, tabagisme, cancer, obésité) |
|
|
Que peut arriver avec un thrombus? Quel est son sort? |
- Propagation - Embolisation - Dissolution - Organisation et recanalisation ( du temps est nécessaire pour rétablissement d'un bon flot) |
|
|
2 types de thrombose, différences. |
1) Veineuse: Peuvent causer de la congestion locale, de l’enflure, douleur ou une tension, mais peuvent aussi s’emboliser. (coeur droit et pulmonaire)
2) Artérielle ou cardiaque: L’athérosclérose joue souvent un rôle important. S’emboliser au cerveau, reins et à la rate (riche apport sanguin).
*thrombus muraux existe aussi (chambres cardiaques ou lumière aortique) |
|
|
Que peut-on déduire des lignes de Zahn des thrombus. |
Ces pâles plaquettes + fibrine et couches foncées de globules rouges furent formées dans le sang et donc pas après la mort |
|
|
Différence entre embolie artérielle et thrombose |
L’embolie artérielle va mener à une nécrose tissulaire alors qu’une thrombose va davantage mener à un œdème.
*Clinique: n’oublions pas que les AINS diminuent l’agrégation plaquettaire. |
|
|
Heparin-induced thrombocytopenia (HIT) syndrome |
Anticorps font que l'héparine ne sert plus à rien comme anticoagulant. Consommation parallèle des plaquettes... |
|
|
Antiphospholipid antibody syndrome : |
Anticorps dirigés contre antigènes de phospholipides prothrombose.
Logiquement, in vitro, inhibe coagulation. Lupus anticoagulant
Mais in vivo, par insultes endotheliales ou par des facteurs directement atteints: Hypercoagulation |
|
|
Coagulation intravasculaire disséminée (DIC) Expliquez globalement physiopathologie. |
Par des micros-thrombus partout dans la circulation, un mécanisme fibrinolytique se met en branle et entraîne consommation des plaquettes, de fibrine et des facteurs.
*susceptibilité hémorragique Mais aussi *susceptibilité hypoxique et hémolytique (thrombus) |
|
|
2 mécanismes enclanchant le DIC |
1) Libération de tissue factor (facteur III) par une destruction massive de tissue, une infection ou blessure endothéliale
2) Lésions répandues de cellules endothéliales= aggrégation plaquettaires *tissue factor aussi* |
|
|
Différence entre embole et thromboembolique |
Composition de la masse intravasculaire:
-embole:gouttes de matière grasse, bulles de nitrogène, débris d’athérosclérose, fragments de tumeur, moelle épinière, corps étrangers, etc. tandis que
-thromboembolie est un thrombus détaché. (fibrine, plaquettes) |
|
|
Conséquence classique de l'embolie |
Nécrose ischémique (infarctus) |
|
|
types d'embolie et origine (part 1) |
1) Embolie pulmonaire: artère pulmonaire 2) Thromboembolies systèmiques: thrombose murales intracardiaques venant d'un ancien infarctus. Points d'arrêts multiples 3) Embolies graisseuses moelle: rupture sinusoÏdes et veinules de la moelle (fractures) Mécanique (poumons et cerveau) et biochimique (acide gras dangereux)
|
|
|
types d'embolie et origine (part 2) |
4) Embolies gazeuses: bulles d'air obstruant circulation (chirurgie, blessures, maladie décompression: nitrogène) 5) Embolies du liquides amniotiques: Infusion du liquide amniotique ou de tissu fœtal dans la circulation maternelle via un déchirement de la membrane placentaire ou une rupture des veines utérines |
|
|
V-F L'infarctus est d'origine veineuse |
faux, origine artérielle. -(embolique ou thrombotiques) -parfois vasospame, hémorraghie athéromateuse, compression tumorale, -rares torsion d'un vaisseau, sac herniaire, ou même veineux (si une seule veine efférente)
|
|
|
Infarctus rouges vs Blancs |
-Rouges: nécrose, mais quand même une vascularisation par (occlusion veineuse, double circulation) Hémosidérine
-Blancs: organes avec tissus épais avec terminaison terminale. (pas de vascularisaiton secondaire possible)
* dans les 2 cas, nécrose de coagulation ischémique. suivie d'une cicatrice (sauf cerveau et sa liquéfaction) |
|
|
Vulnérabilité à l'hypoxie des différentes cellules. |
-Neurone: lésion irréversible après 3 à 4 minutes d’ischémie
oCardiomyocyte:mort cellulaire après 20 à 30 minutes d’ischémie
oFibroblaste: viable même après plusieurs heures d’ischémie |
|
|
Définition d'un choc |
État de débit sanguin inadéquat généralisé résultant en des lésions tissulaires secondairement à l’hypoperfusion tissulaire/cellulaire périphérique.
PA< 90 mmHg.
|
|
|
Types ou origines de choc |
-Cardiogénique: baisse volume éjection (infarctus, rupture ventriculaire, arythmie, embolie) -Hypovolémique: hémorragie, vomissement, brulures - Septique : vasodilatation périphérique. Réponse immune (G+) -Neurogénique: déséquilibre sympa vs para. Vasodilat généralisée -Choc anaphylactique: IgE: vasodilatation systémique, perméabilité, hypoxie |
|
|
V-F Il n'est par rare de voir un DIC dans un choc septique |
vrai. Vasodilatation systémique générale endommage cellules endothéliales. (hypercoagulation)
Agent microbien ou endotoxines -activation facteur XII, -activation du complément -leucocytes, neutrophiles, macrophage (inflammation et pro-coagulation = activation aussi du facteur III, moins de TFPI, thrombomoduline et protéine C= DIC
*mais aussi susceptibilité aux hémorragies par concentration des facteurs aux thrombus, mais indisponibilité et digestion à un autre.
|
|
|
Étapes liant inflammation du choc sceptique et DIC. |
- Sepsis ou septicémie: infection dans circulation sanguine -cellules inflammatoires (TLR)= médiateurs -activation des cellules endothéliales -phénotype procoagulateur par molécules adhésion: thrombose générales
*complément aussi participe à l'inflammation. C3a active les cellules endotheliales= Vasodilat, perméabilité, Hypoperfusion et organe failure. |
|
|
V-F Les patients en choc septiques peuvent démontrer un état "diabétique" |
vrai résistance à l'insuline et hyperglycémie *en raison des cytokines pro-inflammatoires.
Fièvre présente aussi..
|
|
|
3 étapes d'un choc |
1) Non progressive: perfusion ok, tachycarde, vasoconstriction périphérique, perfusion rénale 2) Progressive: hypoperfusion tissulaire + acidose (anaérobie lactique) 3) Irréversible: fuite enzymes + aggravation choc |
|
|
V-F Les glandes surrénales sont hypertrophiées suite à un choc septique afin de compenser |
faux, réduction des cell. corticosurrénales. insuffisance en glucocorticoïdes (anti-inf).
*tubules nécrosés dans les reins également
|
|
|
V-F Les chocs septiques peuvent affecter gravement les poumons par l'hypovolémie. |
faux, généralement peu affectés, sauf si la bactérie parvient aux poumons, dans ce-cas, alvéoles endommagés. |
|
|
But de l'inflammation |
réponse protectrice pour éradiquer une infection ou une lésion cellulaire |
|
|
Caractéristiques et survenues d'une inflammation aiguë: |
- Infections, Nécrose tissulaire/ hypoxique , Molécules inflammatoires, Corps étrangers, Réaction auto-immunes -Exsudat rapide: Protéines, leucocytes (neutrophiles) et donc OEDÈME -Rapide, mais courte durée |
|
|
V-F On retrouve moins de lymphocytes, de macrophages, de fibroses et de nouveaux vaisseaux dans les inflammations chroniques |
faux, on retrouve plus de: -lymphocytes, de macrophages, de fibroses et de nouveaux vaisseaux dans les inflammations chroniques
|
|
|
V-F En même temps que les leucocytes tentent d'éliminer l'agent offensant par phagocytose, des mécanismes anti-inflammatoires prennent place. |
vrai |
|
|
Premiers rendus à l'infection |
Neutrophiles. -Ensuite monocytes-macrophages ET Mastocytes (histamine) |
|
|
Réponse vasculaire |
1) Augmentation du débit sanguin: vasodilatation artériolaire: pression hydrostatique augmentée
2) Perméabilité : espaces interendothéliaux: -leucocytes et protéines. Permettant de concentrer leucocytes au niveau de la lésion.
Résultat: Stase, préparation à réponse cellulaire
|
|
|
V-F Un exsudat trop important peut entraîner un stase sanguin |
vrai, par la viscosité, débit sanguin ralentit : congestion et accumulation de neutrophiles sur l'endothélium.
Cell. endothéliales exprimant aussi molécules adhésion pour leukocytes, stase et inflammation. |
|
|
1) À quoi est dûe la rougeur d'une infection
2) Chaleur
3) douleur
|
1) congestion vasculaire
2) vasodilatation superficielle
3) compression des nerfs avoisinants et amplifiée par (PGE2 et bradykinines) |
|
|
Qu'est -ce que la réponse transitoire immédiate? |
Contraction (rétraction) des cellules endothéliales donnant une augmentation des espaces interendothéliaux. (histamine, bradykinine, neuropeptide P)
*courte durée *si longue durée ou dommages: écoulement prolongé retardé. |
|
|
Expliquez phénomènes transcytose: |
Transport de liquide et protéines par vésicules au travers des cellules endothéliales.
Facteur important: VGEF... |
|
|
Pourquoi le débit lymphatique est-il augmenté durant une inflammation? |
Filtrer l'oedème et diminuer l'impact de celui-ci. Prolifération des vaisseaux (chronique surtout)
infection/inflammation lymphatique: -lignes rouges cutanées Lymphagite: vaisseaux Lymphadenitis: noeuds lymphatiques |
|
|
V-F Les neutrophiles, macrophages, basophiles et éosinophiles sont importants dans le processus inflammatoires par la phagocytose. |
Faux seulement les neutrophiles et macrophages. Toutefois, libération de facteurs de croissance commune à tous. |
|
|
V-F Par le flot sanguin, les erythrocytes occupent habituellement le centre des vaisseaux alors que les leukocytes occupent la périphérie. |
vrai |
|
|
Comment les médiateurs provoquent-ils le roulement des leukocytes?
l'adhesion?
|
en favorisant l'expression des molécules d'adhésion: exemples:
-P et E-Sélectine par Histamine/thrombine se lie à Syalil du leuko. -integrines avides par chemokines
-TNF/IL-1 adhésion neutrophile/endothélium - LFA-1, MAC-1 du leuko lie ICAM-I -VL4 lie VCAM-1
|
|
|
Où et comment a principalement lieu la diapédèse? |
-Veinules post-capillaires.
-Liaison CD31 leuko avec PeCAM (Cd31). Importance de CD44
-Stimulation par chémokines. (endogène: cytokines IL-8, complément C5-a, Eicosanoïdes LTB4) ou (exogène: origine bactérienne) |
|
|
V-F Les leucocytes peuvent utiliser leur granulations pour traverser la membrane basale. |
vrai, collagénase
*Ensuite, chimiotaxisme vers site d'infection ensuite selon un gradient de concentration de médiateurs. |
|
|
Variation de la nature des leucocytes selon la durée de la réponse inflammatoire: |
6-24h: Neutrophiles. (plus nombreux, réponse et adhésion)
24-48h: Monocytes-Macrophages: trop courte demi-vie des neutrophiles. Prolifération au tissu infecté |
|
|
Récepteurs possibles sur les leucocytes pour reconnaître l'agent offensant |
-Récepteurs de produits microbiens: TLR, endosomes et membranes
-Récepteurs de type G protein-coupled: Neutrophiles. Migration et production de substances microbicides.
-Récepteurs pour opsonines: phagocytose: IgG, C3 , Lectines
-Récepteurs pour cytokines: Interféron-Y. Activateur de macrophages. |
|
|
3 étapes de la phagocytose: |
1)Reconnaissance et attachement de la particule: (mannose, Scavenger-endocytose, opsonines)
2) Ingestion et création vacuole phagocytaire: (pseudopodes, vacuole, fusion avec lysosome)
3) Dégradation de la particule ingérée: (ROS et RNS =respiratory burst des neutrophiles) (granules et enzymes lysosomales (élastase))
|
|
|
V-F Naturellement, les neutrophiles favorisent la phagocytose avant la dégranulation enzymatiques. |
vrai
*leurs enzymes lysosomales peuvent endommager les cellules adjacentes même s'il existe des anti-protéases dans le sérum sanguin. |
|
|
Qu'est-ce qu'une réaction allergique |
L'hôte réagit intensivement à des stimuli inoffensifs. Réaction inflammatoire exagérée et atteignant l'hôte. |
|
|
Déficits leucocytaires innés (hérités):
1) Adhésion des leucocytes = ?
2) Fontion phagolysosomale= ?
3) Activité microbicide= ? |
1) Infections bactériennes récurrentes
2) pas de fusion de phagosome et lysosome = Syndrome de Chédiak- Higashi
3) Défaut de l'oxydase phagocytaire: Maladie granulomateuse chronique |
|
|
2 amines vasoactives. Particularités? |
Histamine: Surtout mastocytes dans T.C des V.sanguins. ( un peu plaquettes et basophiles) -Perméabilité + vasodilatation
Sérotonine: Surtout plaquette et neuroendocrine -Vasoconstriction, perméabilité, - contact collagène+plaquettes |
|
|
Quelle est l'utilité de la phospholipase A2: |
Utilisé l'acide arachidonique (20C, eicosanoïdes) pour créér: -prostaglandines: PGI2 (dilat, inhib plaquetes) vs Txa2 (contract, plaquettes) PGE2: Dilat, fièvre, LPS PGD2: Perméabilité, vasodilat
-leucotriènes: vasocontrict, bronchospasme, perméabilité
-lipoxines: inhibiteur inflammation |
|
|
Enzymes à la base des:
-Prostaglandines: Masto, Macro, Endotheliales
-Leucotriènes: Leucocytes |
Cyclooxygenase (COX-1, COX-2)
et
Lipoxygénase (5- et 12-) |
|
|
PGD2: particularité? |
Prostaglandine la plus produite par les mastocytes.
-vasodilatation + perméabilité des veinules - chimioattraction pour neutrophiles |
|
|
PGI2: particularités:
vs
TXA2: particularités |
PGI2: Vasodilatation, inhib adhérence plaquettaire
vs
TXA2: Vasoconstriction, + adhérence plaquettaire |
|
|
Produits de lipoxygénase des leucocytes?
utilités? |
Leucotriènes: -chimioattraction leucocytaire -Vasoconstriction, bronchospasmes, perméabilité vasculaire, adhésion et activation leucocytaire
Lipoxines: Neutrophiles + plaquettes= transcellular biosynthetic pathway activation -inhibitrice inflammatoire et recrutement leucocytaire. -Pas de chimiotaxisme et adhésion |
|
|
V-F Le facteur d'activation plaquettaire (PAF) est un vasoconstricteur et bronchoconstricteur absolu, augmente l'adhésion leucocytaire,chimiotaxisme et augmente synthèse de médiateurs (eicosanoïdes).
|
faux, Tous véridiques sauf qu': à faible concentration, PAF est un important vasodilatateur et accentue perméabilité veineuse.
|
|
|
V-F Lorsque les neutrophiles adhèrent à l'endothélium, ils stimulent la production de ROS par les cellules endothéliales. |
vrai |
|
|
V-F L'expression de l'oxyde nitrique est constante par la NO synthase |
faux, oui pour endothélial (eNOS) et neuronale (nNOS), mais pas macrophages (iNOS). Celle-ci est induite par des cytokines.
**effets: -Vasodilatation -Inhibition de la composante cellulaire de l'inflammation: baisse agréagation plaquettaire, moins de recrutement leucocytaire -Bactéricide |
|
|
2 principales cytokines des macrophages: |
TNF: Coagulant, Adhésion, activation et production cytokines
IL-1: après inflammasome, protéase l'active: Adhésion, Activation et proction cytokines, Prolif et synthèse de collagène |
|
|
V-F Les chémokines spécifiques aux neutrophiles sont les CXC (IL-8) |
vrai Les C-C sont pour tous les leucocytes sauf les neutrophiles tandis que la présence d'un A.A (C-X-C) se spécifie aux neutrophiles.
*NB que ces chémokines sont initialement sécrétées par macrophages activés et cellules endothéliales. |
|
|
2 fonctions des chémokines par leur interaction avec GPCR: |
1) Stimuler le recrutement leucocytaire lors d'une réaction inflammatoire
2) Contrôler la migration des cellules à travers les tissus |
|
|
Protéases acides: utilités et particularités: granules azurophiles... |
Actives à un pH faible. Dégrade les débris et les bactéries dans phagolysosome. |
|
|
Protéases neutres: utilités et particularités: |
incluant collagenase, cathepsin pour dégrader composantes extracellulaires: -collagène, membrane basale - couplage des protéines C3-C5 : anaphylatoxines et kinine-like peptide. |
|
|
Neuropeptide P: utilités et particularités: |
Initiation et propagation de l'inflammation et pression artérielle. - Famille des tachykinine: transmission douleur, perméabilité vasculaire, stimulation endocrine. |
|
|
Parmi la cascade du complément. Quelle molécule est responsable du chimiotaxisme? |
C5a |
|
|
Parmi la cascade du complément. Quelles molécules sont responsables de l'inflammation? |
-c3a et c5a. Stimule relâchement histamine par les mastocytes. (anaphylatoxines)
|
|
|
Parmi la cascade du complément. Quelle molécule est responsable de la phagocytose? |
iC3b va agir comme opsonine et promouvoir phagocytose |
|
|
Parmi la cascade du complément. Quelle molécule est responsable de la lyse cellulaire? |
Le complexe MAC. (C5b-C9) |
|
|
Voies d'activation du C3 convertase |
Alternative: Contact molécules microbiennes (endotoxines)
Classique: Ag-Ig. Protéine C1 s'y joint
Lectine: liaison avec le mannose |
|
|
Sur quels systèmes joue le facteur XII.
|
Facteur XII (Hagemann): -Kinine -Complément -Pro-thrombique -Fibrinolytique
|
|
|
Comment la cascade de coagulation et la cascade inflammatoire s'entrecroisent? (3 aspects) |
-Inflammation augmente production de facteur coagulants, rend la surface endothéliale pro-thrombogénique
-Thrombine de la formation d'un caillot active récepteurs de protéases activées (PAR): P-sélectine, entres autres.
-Plasmine de la coagulation joue dans la cascade du complément dans le complexe C3a/c5a
-Facteur XII active kinine et vice-versa |
|
|
V-F Par l'action de kallikréines, les kinines sont transformées en kininogènes. |
faux Kininogènes----> Kinines (peptides vasoactifs) |
|
|
Effets de la bradykinine. |
Douleur, perméabilité, vasodilation, contraction musculaire |
|
|
Médiateurs de vasodilatation: (3) |
-Prostaglandines. -Oxyde Nitrique -Histamine
*Kinines? |
|
|
Médiateurs de la perméabilité: (6) |
-Histamine et sérotonine -C3a-c5a (via histamine) -bradykinine -Leukotriènes -PAF (Facteur activateur plaquettes) -Neuropeptide P |
|
|
Médiateurs du Chimiotaxisme (5) |
-TNF, IL-1 -Chemokines -C3a, C5a -Leukotriène B4 -Produits bactériens : N-formyl methyl peptides |
|
|
Médiateurs de la fièvre (2) |
-IL-a et TNF -Prostaglandines |
|
|
Médiateurs de la douleur (2) |
-Prostaglandines -Bradykinines |
|
|
Médiateurs pouvant causer dommages tissulaires (3) |
-Enzymes lysosomales des leukocytes -ROS -NO |
|
|
3 conclusions possibles d'une inflammation aiguë? |
1) résolution complète: redevient normal 2) Guérison par une fibrose: gros dommages 3) Inflammation chronique: persistance ou défaut de guérison. |
|
|
3 caractéristiques de l'inflammation aiguë |
- dilatation des petits vaisseaux sanguins -diminution du débit sanguin (viscosité) -Leucocytes et fluides en extracellulaires |
|
|
Manifestations d'une inflammation purulente? |
Pus: neutrophiles, cellules mortes par nécrose liquéfiante et oedème.
*bactéries pyogéniques* *collection en abcès* |
|
|
Qu'est-ce qu'un ulcère? |
Excavation au niveau cutanée ou d'un organe due à la desquamation du tissu nécrotique en inflammation. (lésions irréversibles) |
|
|
Quelles sont les composantes de l'immunité innée? |
- Barrières épithéliales - NK (CMH1-inhibiteur vs Molécules néfastes-activatrices - Système du Complément - Macrophages
*importance initiale: IL1, TNF
|
|
|
Comment l'immunité innée interagit-elle avec l'acquise? |
Complement (CD21) pour l'activation des lymphocytes B. |
|
|
L'immunité acquise débute en moyenne après combien de temps de combat avec le système inné? |
peut commencer après 12h |
|
|
V-F Les lymphocytes à la base de la réponse humorale maturent dans le thymus. |
faux, lympho B mature dans BONE marrow, (moelle osseuse) |
|
|
V-F Les lymphocytes à la base de la réponse cellulaire maturent dans le thymus. |
vrai
-Sélection positive pour ceux qui voient le CMH de soi -Sélection négative pour ceux avec trop d'affinité |
|
|
Composition du BCR, ou B cell receptor? |
Complexe majoritairement formé d'anticorps sur la membrane de la cellule prets à reconnaître des épitopes B. (Ag d'agents pathogènes)
*ne pas négliger importance de CD21 dans co-activation (système complément)
Pour réponse T-dépendante: -CMHII et I -Ligand CD40L et B7 pour co-stimulants -CD58 et CD54 pour adhesion |
|
|
Quelles sont les CPAs principales? (cellules présentatrices d'antigènes) |
Cellules dendritiques: (sous-épithéliales, plasmatoïdes, Folliculaire pour 2e réponse)
*Ne pas négliger: activation de CD4 avec feedback positif - Lymphocyte B - Macrophages |
|
|
V-F Le CMH1 des CPA est très important pour activé CD4 naifs. |
faux, CMH2, utilisation des épitopes T (fragmentation de molécules exogènes) |
|
|
Quel est l'utilité des molécules B7 des CPAs? |
Contrôle la migration et stabilise le complexe avec TCD4 par le CD28 |
|
|
Quel est l'utilité du CD40 des TCD4? |
Reconnaissance et liaison stable avec cellules désirées (Lympho B, Macrophages) |
|
|
Par quel moyen les TCD4 stimule les lymphocytes B? |
Par les interleukines (cytokines), IL-2 précisément.
N.B aussi sécrétion de TNF-B et IFN-a par Th1 |
|
|
V-F Le BCR est utile pour reconnaître cytokines dans l'activation des lympho B |
faux, BCR utile pour détection des épitopes B, -Polysaccharides, lipides= IgM surtout
|
|
|
Qu'est-ce que l'immunité passive? |
Présence d'une défense immunitaire par IgG maternelles dans le corps du nourrisson. |
|
|
V-F Au lieu de faire un plasmocytes exécutif immédiatement, le lymphocyte B actif peut se différencier en lymphocyte B mémoire |
vrai |
|
|
V-F Les IgA utilisent un effecteur secondaire pour neutraliser l'agent pathogène par son antigène. |
Faux, neutralisation directe dans la lumière des muqueuses. (g-i, urinaire) |
|
|
V-F autant que les anticorps peuvent activer la cascade du complément, autant qu'ils peuvent "opsoniser" directement. |
vrai |
|
|
Les TCD8 peuvent être activés par les TCD4, mais quel est l'autre moyen? |
Directement par le CMH1 d'une cellule lésée et par détection de la protéine endogène déficiente. |
|
|
V-F Les lymphocytes T mémoires sont présents en quantité négligeable. |
faux, plus abondantes que les TCD4 naives.
Auto-Stimulées par IL-2 |
|
|
Comment se nomment les lymphocytes T effecteurs et destructeurs finaux ? |
CTL (Lymphocytes T cytotoxiques)
Reconnaissance par CMH1 d'une cellule lésée |
|
|
Comment les TCD4 activent-ils les macrophages? |
Les lymphocytes Th1 sécrètent le médiateur IFN. |
|
|
Comment les TCD4 activent-ils les neutrophiles, importants pour la réponse inflammatoire? |
Après avoir été différenciés (TGF-B, IL-6, IL-23)
Les lymphocytes Th17 sécrètent les médiateurs IL-6 (chimiotaxisme) et IL-17. |
|
|
Comment les TCD4 activent-ils les Mastocytes, IgE et eosinophiles dans la réponse aux parasites? |
Après avoir été différenciés (IL-4) Les lymphocytes Th2 sécrètent les médiateurs: -IL-4, qui accentue les IgE des lymphos B. et Mastocytes -IL-5: éosinophiles -IL-13: Mucus |
|
|
Les 2 systèmes utilisés par les TCD4 pour activer les autres cellules |
1) Cytokines 2) CD40L |
|
|
Que sont les follicular helper T cells., |
Migration dans les follicules des organes lymphoïdes où ils furent activés pour assurer la stimulation des réponses des anticorps |
|
|
Pourquoi parle-t-on de mécanisme d'activation cellulaire T-indépendant? |
passe directement par des épitopes "B" de polysaccharides vers la reconnaissance des IgM |
|
|
Pourquoi parle-t-on de mécanisme d'activation cellulaire T-dépendant? |
Doit passer par les TCD4 pour reconnaître un antigène (épitope T).
*N.B: Le lympho B peut être le CPA, activé un CD4 par CHM2, et recevoir les cytokines pour différencier en plasmocyte et produire les IgG |
|
|
Qu'est-ce que le phénomène d'heavy-chain class (isotype) switching? |
Different antibodies for different functions, but still the same specificity.
Aussi, plus grande affinité (chaine lourde variable) fait que les IgM deviennent IgG. |
|
|
Qu'est-ce que le phénomène d'affinity maturation? |
Stimulation d'une production d'anticorps avec une très forte affinité à l'antigène de l'agent pathogène.
*à l'origine de "B-memory cells" |
|
|
V-F Le but de la vaccination est d'assurer une production d'IgM constante. |
faux, on cherche des cellules B mémoires avec IgG à forte affinité pour une protection à long terme.
|
|
|
Pourquoi y a-t-il une grande variabilité dans les HLA ou CMH? |
Pour un loci sur le chromosome 6 plusieurs haplotype: 6 sortes pour CMH1 et plus de 20 pour CMH2 |
|
|
Comment sont éliminés les lymphocytes |
apoptose |
|
|
V-F Les cytokines, polysaccharides, sont très utilisés par les TCD4 |
faux, les cytokines sont des polypeptides.
*Pleiotropisme vs redondance* |
|
|
Donnez trois exemples de maladies où l'on retrouve un stimulus persistant (voir, inflammation chronique.
|
-Les infections persistantes (ex. tuberculose),
-l'exposition à du matériel étranger non-dégradable (ex. accumulation pulmonaire de particules de silice = silicose),
-les maladies auto-immunes (ex. lupus érythémateux, où l'individu forme une réaction immunitaire contre lui-même)
-microbien: syphillis |
|
|
Trois événements surviennent simultanément dans un foyer d'inflammation chronique. Quels sont-ils? |
- Mononucléées cells (tout sauf neutrophiles) - destruction par stimulus ou inflammation. - Réparation Angiogénèse et fibrose (collagène) |
|
|
Nommez les différents types de cellules qu'on peut retrouver dans un site d'inflammation chronique. Décrivez brièvement. |
-Macrophage : destruction des microbes et phagocytose; sécrétion de produits qui augmentent la destruction tissulaire, la prolifération vasculaire, la fibrose et la réaction immunitaire.
-Lymphocytes : réactions immunitaires avec production d'anticorps, réactions cellulaires immunes ou non-immunes, production de cytokines pour activer les macrophages.
-Plasmocytes : production d'anticorps.
-Eosinophiles : phagocytose, production de facteurs pouvant détruire des parasites.
-Neutrophiles : destruction de microbes, production d'enzymes. PAS BCP PRÉSENTS
-Fibroblastes : synthèse de collagène.
|
|
|
Expliquez la différence entre un monocyte, un macrophage et un macrophage activé. |
-dans le sang, il s'agit de monocytes. -dans les tissus, elles deviennent des macrophages, cellules plus grosses que les monocytes et capables de phagocytose. -Après stimulation appropriée (entre autres par des produits sécrétés par les lymphocytes), les macrophages deviennent plus gros, augmentent leur activité métabolique, leur capacité à la phagocytose, la destruction des microbes et la sécrétion de produits biologiquement actifs. Il s'agit alors de macrophages 1 activés. (IFN-alpha) ou M2 activés (Il-4,Il-13) |
|
|
Décrivez trois mécanismes pouvant expliquer l'augmentation du nombre de macrophages dans un site d'inflammation chronique |
1- recrutement continu de nouveaux macrophages provenant de la circulation sanguine 2- prolifération locale de macrophages 3- immobilisation des macrophages qui sont déjà sur place, empêchant leur départ |
|
|
Le macrophage est considéré comme la cellule la plus importante dans l'inflammation chronique à cause des multiples rôles qu'il peut jouer et des nombreux produits qu'il peut sécréter : |
1.phagocytose des substances toxiques, irritants ou agents infectieux 2.sécrétion de produits causant de la destruction des tissus (MMPs et élastase) 3. sécrétion de produits causant de la fibrose (PDGF, FGF2, TGF-B) et angiogénèse (NO, VEGF, FGF2 et ANG-1 pour stop)
4.joue un rôle dans les phénomènes inflammatoires non immuns mais aussi dans la présentation des antigènes au début de la réaction immunitaire, et dans la phase effectrice de l'immunité cellulaire (hypersensibilité retardée IVa) |
|
|
Quels sont les deux types principaux de granulomes? Mentionnez quelques étiologies possibles pour chacun des types. |
-granulomes à corps étranger: se forme autour de corps étrangers inertes, par exemple des fils de suture en matière synthétique. La formation de ces granulomes ne représente pas une réaction immunitaire contre ces corps étrangers.
-Les granulomes immuns, par contre, résultent de l'activation des macrophages dans le cadre d'une réaction d'hypersensibilité retardée. On peut les retrouver dans plusieurs types de maladies, infectieuses ou non. Un exemple classique de maladie granulomateuse est la tuberculose. La séquence des événements impliqués est celle de la réaction d'hypersensibilité retardée IVa.
|
|
|
Quels sont les différents types de cellules qu'on peut retrouver dans un granulome ? |
-petits macrophages -macrophages à cytoplasme abondant (« épithélioïdes) -macrophages multinucléés -lymphocytes -parfois plasmocytes et neutrophiles
*Fibroblastes* ceinturant pour faire capsule |
|
|
Granulome à corps étranger. Explication de suture à permanente.. |
Cette situation est un exemple de formation de granulome à corps étranger. Ces granulomes se forment autour de produits inertes que les macrophages sont incapable de digérer. Étant recouvertes de macrophages, ces particules étrangères sont moins dommageables pour les tissus, et peuvent persister pour toute la vie de l'individu.
Des granulomes semblables peuvent se former autour de structures qui ne sont pas vraiment des corps étrangers. Par exemple, on peut en retrouver dans la peau au pourtour de kystes rompus, les macrophages étant alors activés par la kératine contenue dans le kyste et qui est en contact avec le derme. |
|
|
Importante de différencier chronique granulomateuse de chronique non-granulomateuse |
l'identification de granulomes dans un foyer d'inflammation chronique réduit beaucoup la liste des diagnostics possibles et permet dans la plupart des cas d'identifier avec précision l'étiologie de la réaction inflammatoire (ex. tuberculose). D'où l'intérêt des granulomes! |
|
|
Étapes de la fibrose ou tissue repair: |
1- formation de nouveaux vaisseaux (NO, VEGF, FGF2) et stop angiogénèse (PDGF, TGF-B, ANG-1) 2- migration et prolifération des fibroblastes (PDGF, TGF-B, FGF2) 3- production de matrice extracellulaire (FGF2, PDGF, Il-1, IL-13 4- maturation et organisation du tissu fibreux (TIMPs) |
|
|
Plaie chirurgicale linéaire, ses rebords sont nets et l'opération est faite avec des techniques stériles . Comment sera la guérison? |
Plaie qui devrait guérir par première intention. On peut s'attendre à une re-épithélialisation en 24 à 48 heures, période durant laquelle la plaie est infiltrée par des neutrophiles et des macrophages. Par la suite, le tissu de granulation formé devrait être peu abondant, et la cicatrice devrait guérir assez rapidement sans laisser de séquelle majeure. Il est toutefois illusoire de penser que rien ne paraîtra. |
|
|
Type de guérison d’un ulcère d’estomac! |
La guérison d'un ulcère d'estomac est un exemple de guérison par seconde intention. La nécrose des tissus est abondante, et la guérison nécessite la formation d'une quantité importante de tissu de granulation. Ce tissu se forme à partir du tissu sain autour de la plaie, et a une architecture très bien définie. En surface, on retrouve un exsudat de fibrine et de neutrophiles; dans la portion moyenne se trouve une importante prolifération de petits vaisseaux sanguins fragiles, à paroi mince et peu étanche, le tout accompagnés d'œdème et de cellules inflammatoires mononucléées. Enfin, la portion profonde est occupée par une prolifération de fibroblastes synthétisant du collagène. |
|
|
Pourquoi laver et décontaminer une plaie? |
Une plaie contaminée par des bactéries et des corps étrangers va guérir moins vite qu'une plaie propre. Dans une plaie contaminée, la phase initiale d'infiltration de la plaie par des neutrophiles et macrophages sera plus marquée que dans une plaie stérile étant donné qu'il y a un stimulus persistant qui active continuellement la réponse inflammatoire. Le système immunitaire peut réussir à se débarrasser des bactéries et la guérison peut alors suivre son évolution normale, mais au dépend d'une plus grande perte tissulaire et d'une cicatrice plus importante. Il est préférable d'aider la guérison en enlevant le tissu nécrotique (débridement) et en lavant la plaie |
|
|
Pourquoi ne pas refermer une paie infectée purulente? |
Présente des signes d'inflammation aiguë, et laisse s'écouler du pus. On ne doit pas refermer une telle plaie étant donné qu'il faut dans un premier temps traiter l'infection avant que la guérison ne puisse se produire. La plaie doit rester ouverte pour permettre l'écoulement du pus. Vous pourrez ajouter un traitement aux antibiotiques. Si vous refermez la plaie, vous créez un milieu propice à la prolifération bactérienne, et la destruction tissulaire sera plus marquée, avec formation d'une cicatrice plus importante. |
|
|
Vitamine C vs Scorbut |
- La déficience en vitamine C perturbe la synthèse du collagène et le rend moins résistant.
-La vitamine C joue un rôle important dans la synthèse du collagène, étant impliquée dans l'hydroxylation de la proline et la lysine. L'hydroxylation de ces deux acides aminés est nécessaire pour la liaison des chaînes alpha et pour le "cross linking" des molécules de collagène, ce qui lui donne sa résistance.
-Il en résulte une faiblesse par exemple des vaisseaux sanguins, ce qui entraîne des saignements. On peut encore voir de nos jours des cas de scorbut chez des alcooliques qui ne se nourrissent essentiellement que d'alcool et qui ont des déficits marqués en vitamines. |
|
|
Expliquez les principes de la force tensile des cicatrices.
|
- 24-48h: collagène, prolif épithéliale - 1 semaine : 10% de la force tensile de la peau normale - 4 semaine : grosse progression -3 mois: 70-80%
On peut enlever les points de suture après une semaine, mais le patient doit faire attention aux efforts violents pour plusieurs semaines. |
|
|
Explications Infarctus passé VS capacité contractile, dans un contexte d'insuffisance du coeur gauche. |
L'infarctus a guéri par seconde intention en formant une large cicatrice. Il s'agit toutefois d'un tissu de remplacement, qui ne possède pas les propriétés fonctionnelles du tissu initial, en particulier le myocarde dans ce cas. La cicatrice ne peut donc pas pomper le sang et il en résulte que le cœur est très hypothéqué, ne pouvant fonctionner adéquatement avec le myocarde résiduel, ce qui entraîne les symptômes du patient |
|
|
Structure d'un tissu de granulation lors de la guérison d'une plaie? |
Apparition de 24 à 72h après début réparation Visuellement, le tissu de granulation présente une couleur rose et une apparence granuleuse. D’un point de vue histologique, on note la formation de nouveaux vaisseaux sanguins « leaky » et la présence de fibroblastes. De plus, il est souvent œdémateux. |
|
|
Quelle est la protéine principale constituant une cicatrice?
|
Le collagène. |
|
|
Une plaie infectée par des bactéries va-t-elle guérir plus vite qu'une plaie stérile? |
Non. -Une plaie stérile fera intervenir le mécanisme de réparation tissulaire mais non celui de réponse inflammatoire (absence d’agents / facteurs microbiens). -Cependant, une plaie infectée devra faire intervenir les 2 mécanismes. Et tant et aussi longtemps que l’agent microbien ne sera éliminer, la réponse inflammatoire continuera; il y aura donc toujours des cellules qui mourront, généreront du peut-être du pus et devront être remplacées. |
|
|
DTH ou hypersensitivité retardée? qu'est-ce? |
Réaction immunitaire au cours de laquelle l’activation des macrophages par les lymphocytes T et l’inflammation provoquent des lésions tissulaires chez un individu ayant déjà été exposé à l'agent.
ex: tuberculose. |
|
|
V-F Les T-CD4 assure des fonctions effectrices par leurs cytokines et par leur récepteur CD40. |
faux, ils ont le ligand CD40L, les macrophages et lymphocytes B ont le récepteur |
|
|
Macrophages tissulaires différenciés: plusieurs noms selon localisation? |
o Microglie dans le SNC o Ostéoclastes dans les os |
|
|
Composition radiale (du centre vers l'extérieur) d'un granulome |
Inflammation chronique avec présence de macrophages par remplacement des neurophiles en 2-3 semaines:
macrophages epithelioid < multinucleate giant cells< lymphocytes < fibroblastes et zone fibreuses |
|
|
2 moyens qu'ont les CD8 pour être cytotoxiques. |
1) Système perforine-granzymes = caspases= apoptose
2) FasL du CD8 au récepteur Fas sur la cellule infectée. |
|
|
Cytokine(s) pour auto-prolifération des lympho T |
IL-2 |
|
|
Cytokine(s) pour différenciation des lympho Th1 (macrophages) |
Il-12, IFN-alpha, - Il-18
sécrétion ensuite de: -Il-1 (neutrophiles) -Chémo -TNF (inflammation) -IL-12 (amplification Th1) |
|
|
Cytokine(s) pour différenciation des lympho Th17 (neutrophiles) |
IL-1, IL-6 et IL-23.
-sécrétion ensuite d'IL-17, IL-22 (chimioattractive) pour neutrophiles
-auto-amplification par IL-21 |
|
|
Quel type de nécrose s'apparente au foyer central des granulomes |
nécrose caséeuse |
|
|
Les T-CD8 reconnaissent les CMHII par leur TCR |
faux, CMHI, pas le CMHII |
|
|
V-F Les T-CD8 produisent aussi IFN et pourraient s'apparenter à la réaction d'hypersensibilité retardée. |
VRAI |
|
|
Maladies présentant des granulomes |
la tuberculose, la lèpre, la syphilis, la maladie du cri du chat, la sarcoïdose et la maladie de Crohn |
|
|
4 Types d'hypersensibilité et leur voie principale |
Type I: Allergie, Hypersensibilité immédiate Type II: Ig-Ag interaction Type III: Complexes immuns Type IV: Hypersibilité par médiation cellulaire |
|
|
Pour les réactions allergiques: Que veut-on dire par atopique? |
Les personnes qui ont une forte propension à développer ce type de réactions.
Influence de la génétique |
|
|
2 phases de l'allergie. (Type I) |
Immédiate: 5 à 30 min jusqu'à 1 heure = vasodilat, leakage, contraction muscle lisse et/ou sécrétion glandulaire. (histamine, Leukotriènes)
Tardive: de 2h à 24h après = infiltration par éosinophiles, neutrophiles, basophiles, monocytes et lympho T = destruction tissulaire |
|
|
V-F Les basophiles sont similaires aux mastocytes, tant par leur composition que leur quantité dans le sang. |
faux, même FCreceptors pour IgE et granulations,
mais beaucoup moins présent dans le sang |
|
|
Importance des Th2 dans l'hypersensibilité type I par cytokines produits: - Il-4= - Il-5= - Il-13 |
-IL-4 =sur cellules B pour commutation isotypique pour IgE
- IL-5= Activation eosinophiles
- IL-13: mucus
*ne pas négliger feedback positif par chimiokines sur Th2
|
|
|
V-F Un mastocyte sensibilisé est celui capable de synthétiser des IgE à plusieurs allergènes |
faux, il peut se lier à plusieurs IgE spécifiques à plusieurs allergènes par leur Fc, mais ceux-ci sont synthétiser par lympho B.
*sensibilisé à une activation par une rencontre ultérieure qui prend 7-10 jours* |
|
|
3 moyens d'actions des mastocytes sensibilisés lors du 2e contact avec antigène. |
* Dégranulation des médiateurs préformés 1)= amines vasoactifs, enzymes, protéoglycans) 2)=ECF-NCF pour late phase neutro,eosino
|
|
|
Actions du PAF (facteur d'activation plaquettaire) |
* Aggrégation plaquettaire
N.B : petite dose= vasodilatation |
|
|
Manifestation d'anaphylaxie systémique: |
* Choc vasculaire |
|
|
Exemple de réaction locales d'hypersensibilité immédiate |
Fièvre, asthme, urticaire par exposition localisée à un allergène commun |
|
|
Interleukine à l'origine de rhinite et sinusite |
Il-13 produite par Th2= hausse de la sécrétion de mucus |
|
|
Dans l'hypersensibilité de type II, par quels moyens les anticorps altèrent-ils les cellules? |
1) Opsonisation ( Directe IgG, activation classique complément, ou citotoxicité tissulaire)
2) Inflammation: C3a,C5= perméabilité et chimiotaxisme + protéase. ADCC (NK, neutro, éosino= ROS)
3) Fctnnement cellulaire: inhibition de récepteurs par anticorps (négativement dans le muscle (Ach), positivement pour la thyroïde (TSH)) |
|
|
Exemples d'hypersensibilité de type II avec phagocytose pathologique |
* Réactions de transfusion = les cellules incompatibles du donneur sont opsonisées par des anticorps préformés.
* Maladie hémolytique du nouveau née= les anticorps de la mère (igG) traversent le placenta et s’attaquent aux globules rouges de l’enfant.
* Réactions à des médicaments= le médicament vient s’attacher à la surface des globules rouges et les anticorps sont produit contre le complexe protéine-médicament dans la membrane. |
|
|
V-F Les gros complexes immuns sont les plus dangereux |
Faux, Les petits complexes avec un excès d'Ag sont plus suspects de persister aux attaques des macrophages.
Tous les sites Fc des Ig sont occupés. |
|
|
Où tendent les complexes immuns à se déposer? |
* dans les vaisseaux sanguins au niveau des sites de turbulence(ramifications des vaisseaux) ou de pression élevées ( glomérules rénaux ou synoviaux) |
|
|
V-F La formation de complexe immun est immédiate |
faux, habituellement après la formation d'Ac: qui prend une semaine. |
|
|
Par quel mécanisme les complexes immuns causent-ils des dommages? |
Réaction inflammatoire aïgue. (complément + neutrophiles)
(fièvre, urticaire, arthralgies, adénopathies, protéinurie) = plaquettes activées = facteur d'Hageman = thrombine = ischémie qui empire le cercle vicieux de la vasculite. |
|
|
V-F Une hausse marquante de C3 sérique témoigne d'une grande activation du complément et de l'inflammation par les complexes immuns |
faux, on aura une consommation, donc un niveau diminué des protéines du complément |
|
|
Types d'inflammation par complexe immuns selon localisation: vaisseaux, rein, articulation |
Vasculite, glomérulonéphrite, Arthrite |
|
|
Dans quel contexte peut-on avoir un complexe immun local? |
Quelqu'un d'immun avec un excès d'anticorps préformés.
Par injection de l'antigène localement, il y aura rapidement un complexe par l'arrivée des Ig préformés.
Présence d'oedème locale et hémorragie, pouvant évoluer en ulcère par nécrose dans le vaisseau. (fibrinoid necrosis) |
|
|
V-F Tous les complexes immuns sont dommageables |
faux, la majorité sont directement phagocytés par l'affinité des macrophages à la chaîne lourde des Ig |
|
|
Interleukine utile à la prolifération des CD4+ naïfs lors d'une primo-exposition par un CPA |
IL-2
(facteur de croissance autocrine) |
|
|
Utilité ou actions d'IFN-gamma sur macrophage |
- haute capacité de phagocytose -haute expression de CMHII - haute sécrétion de TNF, IL-1, et chimiokines et donc inflammation -IL-12 donc prolifération |
|
|
Sécrétions et actions des Th17 |
- IL-17 et 22: neutrophiles et monocytes
- IL-21: auto-activation des Th17 |
|
|
V-F La DTH (hypersensibilité retardée) est caractérisée par une accumulation de neutrophiles |
faux, davatage les macrophages (Th-1).
à long terme, peut devenir granulomateuse (avec epithéloïde, giant cells, lymphocytes et fibroblastes) |
|
|
Maladies systémiques relié à l'hypersensibilité de type IV, ou (cell=mediated). |
- Diabète type I (aussi avec CTLs) - Crohn disease - Rejets transplantations etc.. |
|
|
V-F Dans certaines réactions d'hypersensibilité retardée, la sécrétion d'IL-2 par les cellules CD4+ active les cellules T CD8+ cytotoxiques. La cytotoxicité médiée par les cellules T est donc une extension de la réaction d'hypersensibilité retardée. |
VRAI |
|
|
Pourquoi dit-on que les Lympho T ont une double spécificité? |
le récepteur des Lym T (TCR) reconnaît certains résidus d’antigènes peptidiques, mais reconnaît également les résidus polymorphes de la molécule du CMH qui présente ce peptide. |
|
|
Par rapport à la structure de CMH1, la chaine-alpha extracellulaire est divisisée en 3 domaines. Quelles sont leur utilité respective? |
1 et 2: site de liaison en forme de sillon au peptide provenant d'une protéine cytosolique
3: site de liaison au corécepteur CD8 des lympho-T. |
|
|
Par rapport à la structure de CMH2, les chaines alpha et béta extracellulaire sont divisisées en 2 domaines chaque. Quelles sont leur utilité respective? |
-alpha 1 et Beta 1: goutière pour peptides
-B2: corécepteur CD4 |
|
|
V-F La défense contre les microbes cytoplasmiques est bien effectuée par la voie humorale. |
faux, cytoplasmique= intracellulaire= voie cellulaire (Cd8 et CTL) |
|
|
Quel est l'objectif des cytokines hématopoïétiques? |
Leur fonction est d’augmenter le nombre de leucocytes lors d’une réponse immunitaire et inflammatoire et de renouveler les leucocytes qui ont été perdus lors de telles réponses. |
|
|
V-F alloantigène et xénoantigène sont synonymes |
Faux,
allo: différence intra-espèce
xéno: pas la même espèce |
|
|
Pourquoi, par la reconnaissance des molécules d'un CMH allogénique, on peut parler d'une réaction immunitaire croisée?
qu'elle principe détourne-t-on ce faisant? |
un lympho T va percevoir un CMH étranger comme si c'était un CMH du soi, mais avec un peptide étranger.
CMH Restriction non respectée. Pas sensé reconnaître des CMH du non-soi. |
|
|
Que sont les antigènes mineurs d'histocompatibilité? |
Antigènes non CMH qui induisent un rejet de greffe.
Protéines cellulaires normales, mais différentes entre donneur et receveur. (transfusion et transplantation de moelle) |
|
|
Voie directe vs Voie indirecte
Différences? |
Directe: lympho T détecte le CMHI et CMHII du CPA étranger. (DTH ou CTLs). Rejet aïgu cellulaire ou humoral
Indirecte: CPA du soi, procède le CMHII allogénique et le présente comme Ag au lympho T. réaction DTH. Chronique.... |
|
|
2 réactions de rejet médiées par immunité cellulaire |
- Hypersensibilité retardée (macro, neutrophiles) par CD4 (Th1, Th17)- Réaction chronique
- Cytotoxicité: Cd8 === CTLs (Aiguë cellulaire) |
|
|
Si le rejet de greffe se fait par une réaction humorale, à quoi les anticorps vont-ils principalement s'attaquer? |
La cible initiale de ces anticorps semble être la vascularisation du greffon. -si préformés par exposition ultérieure= hyperaïgue - sinon= Aïgue
*Réaction fidèle à l'immunité humorale. (Indirecte) |
|
|
Le rejet hyperaigu (quelques minutes) est associé à quel type de réaction. |
Humorale. Quelques minutes plus tard, avec anticorps circulants pré-formés. Attaque la vascularisation de la greffe.
(se lient aux antigènes sur l’endothélium vasculaire du greffon, active le complément par la voie classique et de la coagulation pour provoquer des lésions de l’endothélium et la formation d’un caillot = nécrose et thrombose |
|
|
Le rejet aigu (jours/semaines) est associé à quel type de réaction. |
Cytotoxique médié par CD8-CTLs: -quelques jours à semaines... plus communes des échecs précoces
CTL contre vaisseaux ou cellules constitutives du greffons. Exemple: renal failure et endothélite
*participation également d'une réaction humorale aigüe
|
|
|
Le rejet chronique (mois/années) est associé à quel type de réaction. |
T-cell par macrophages et neutrophiles. (DTH). Cause principales des échecs de transplantation: mois ou années...
-Fibrose du greffon et athérosclérose des vaisseaux. - CD4 mediated: cytokines stimulant fibroblastes, cellules musculaires lisses, éosinophiles, neutrophiles. |
|
|
V-f Les tumeurs malignes sont bien différenciés. |
faux, elles sont peu différenciés, mais ont une plus grande capacité de prolifération et d'envahissement que les bénignes. |
|
|
Définitions:
1) Séminome:
2) Lymphome:
3) Harmatome:
4) Choristome:
5) Tératomes: |
1) Séminome: Germinome, des testicules
2) Lymphome: provenant de lymphocyte
3) Harmatome: masse désorganisée indigène bénigne (cartilage dans poumons)
4) Choristome: anomalie congénitale, reste cellulaire hétérotopique
5) Tératomes: cellules totipotentes |
|
|
V-F Sarcomes et carcinomes sont des tumeurs bénignes. |
Non! Malignes. -Sarcome (tissus) -Carcinome (épithéliale de mésoderme, ectoderme ou endoderme)
*adéno si glandulaire, transitionnelle (vessie) , épidermoïde ou squameux
|
|
|
distinction entre cellules néoplasiques malignes et bénignes (4 critères) |
1) différenciation faible: anaplasie= maligne 2) taux de croissance= élevé = maligne 3) invasion locale= pas de capsule fibreuse 4) Métastases= tumeur maligne
|
|
|
V-F Une dysplasie est irréversible |
faux, elle peut redevenir normale et regagner une certaine polarité. Ne progresse pas toujours en cancer.
Si c'est le cas, carcinome in situ (confinement dans la membrane basale, maligne non-dissiminée) |
|
|
V-F Les cellules tumorales ne traversent pas leur cycle cellulaire plus rapidement que les cellules normales. |
vrai |
|
|
3 moyens de propagation en métastases des cellules tumorales: |
- Cavités corporelles: - réseau lymphatique: nodule sentinelle. Par carcinome - réseau vasculaire: sarcomes par veines surtout et fini vers foie ou poumons habituellement. |
|
|
V-F Environ 30% des cancereux ont une prédisposition génétique |
faux, moins de 10% |
|
|
Héritage autosomique dominant. Explication |
Une chance sur 2 d'obtenir le gène muté de notre parent. Si on l'obtient, on perd un allèle du gène répresseur de tumeur. Une seule mutation nécessaire pour désactiver le gène. c'est donc une "transmission dominante" d'une mutation récessive.
- rb: rétinoblastome - p53 - APC: polypes colon - BrCa1,BRCa2: seins, ovaires
*N.B: une seule perte allélique, par haploinsuffisance, peut être nécessaire.
|
|
|
Meilleur exemple de cancer par héritage autosomique récessif. (réparation de l'ADN) |
Xeroderma pigmentosum. (excision des nucléotides inefficace) |
|
|
Que veut-on dire par sélection tumorale? |
Les tumeurs sont de plus en plus aptes à résister à l’hôte plus le temps avance= -le moins antigéniques -le moins de facteurs de croissance
progession tumorale: plus en plus aggressives et malignes |
|
|
Exemple de mutation pouvant mener à l'expression d'un proto-oncogène, voir un oncogène. |
amplification génique mutation ponctuelle translocation chromosomique
*miRNA, épigénétique* |
|
|
Phénotype mutateur. Qu'est-ce? |
Cellule dont la réparation de l'ADN est mise en péril. Accumulation rapide de mutation.
(BrCA1,BrCA2, LMH1, MSH2) |
|
|
8 altérations fondamentales menant à l'adoption d'un phénotype malin |
1) Autosuffisance de Growth factors 2) Insensibilité aux inhibiteurs de croissances 3) Évitement apoptose 4) Réplication illimité 5) Angiogénèse 6) Métastase 7) Réparation de l'ADN déficiente 8) Évitement du système immunitaire |
|
|
Répercussions possibles d'une oncoprotéine |
1) Facteur de croissance mutés (surexprimé) 2) Récepteurs à GF (tyrosine-kinase constamment dimérisée) 3) Protéine transductrice (GAP, laisse RAS activée avec GTP, voie MAP-kinase) 4) tyrosine kinase cytosolique activée (faux-récepteur) : BCR-ABL cross-talk 5) Facteurs de transcription pour prolif (MYC) 6) CDK- cyclines , diminution inhibition. |
|
|
Évolution des complexes CDK/cyclines |
- CDK4-cycline D: RB, E2F libre, checkpoint G1/S -CDK6-cycline D: après checkpoint G1/S - CDK2-cycline E fin G1 -CDK2-cycline A: durant S, vers checkpoint G2-M -CDK1-cycline A -CDK1 -cycline B : G2/M transition
*inhibition générale par: p.21,27,57 *inhibition de CDK4: p16,15,18,19 p53, TGF-B
|
|
|
V-F MDM2 tend vers la prolifération cellulaire.
|
Vrai, anti-p53, qui lui, est le principal frein dans le checkpoints du cycle cellulaire.
ATM empêche dégradation de p53 avec MDM2 |
|
|
Explication du checkpoit G1/S avec RB
(the governor) |
RB est un inhibiteur de croissance. S'il est phosphorylé par CDK4/CD, il libère le E2F qui lui stimulera la transcription des gènes régulant la réplication.
|
|
|
Importance du gène p53 (chromosome 17) comme suppresseur de tumeur |
*Li-fraumani syndrome si 1 allèle mutée.
facteur de transcription : - arrêt du cycle temporaire par p21 -réparation GADD45 -Senescence par mir45, - Apoptose (Bax, Puma)
auto-destruction par MDM2 si réparation terminée.
|
|
|
Gène APC et polype adénomateux du colon. Description du phénomène |
APC est responsable de la dégradation de B-caténine. Sinon, B-caténine active TCF et la prolifération cellulaire par MYC
*mutation d'APC le rend inactif. donc prolifération constante.
*même principe pour E-cadhérine de contact non-fonctionnel, qui relâche les B-caténines. (mécanisme à l'origine des blessures) |
|
|
Actions importantes des gènes suppresseurs de tumeurs:
-INK4/ARF (CDKi/inhibe MDM2) - TGF-B - PTEN - NF1 - NF2 - VHL - WT1 - patched
|
-INK4/ARF (CDKi/inhibe MDM2) - TGF-B: active p21 (CDKi) par SMAD - PTEN: inhibe (AKT,PI3K) - NF1: GTPase = inactivation de RAS - NF2: neurofibromine 2 active E-cadherine. - VHL: HIF-alpha pas dégradé: prolifération même si pas hypoxique - WT1: pour différenciation, anti-prolif - patched: régulation de TGF-B |
|
|
À partir du moment où les dommages cellulaires sont trop importants, quel est la cascade intrinsèque apoptotique |
p53 active Puma (BH3), qui inhibe BCL2, qui permet expression de Bax/Bak, qui permet d'augmenter perméabilité mitochondriale: -libération de citochrome C, activation d'APAF1 -caspase 9 activée, active caspase 3 executrice
*N.B inhibition possible de caspase 9 par IAP |
|
|
V-F La cascade apoptotique extrinsèque, avec Fas/FasL et dead domain. Permet activation des caspases-9 et ensuite, de caspase exécutrice 3. |
faux, caspase-8
*N.B: FLIP permet d'empêcher activation des caspases 8. |
|
|
Quel est le seul moyen pour une cellule ayant perdu le contrôle de ses réplications, (Bridge-fusion-breakage cycle) de sortir du cycle tout en évitant la catastrophe mitotique. |
Réactiver sa télomérase.
N.B: les caryotypes seront complexes, avec un phénotype malin. |
|
|
Switch angiogénique des tumeurs: 2 facteurs favorisant angiogénèse qui permet la croissance et les métastases |
- HIF1-alpha non dégradé par VHL= cytokines (VEGF + bFGF)
- Protéases: libération de facteurs stockés dans MEC.
Pro-angio: VEGF -ECM vs bFGF *NOTCH= density and branching
Anti-angio : par TSP1 -Plasminogen= angiostatine -Collagen= endostatine -Transthyretin= Vasculostatine |
|
|
Étapes de l'invasion de la matrice extra. |
1) Relachement des interactions (E-cadherines, Caténines) SNAIL/TWIST inhibe E cadherine.
2) Dégradation de membrane et tissu conjonctif (MEC)- MMP et autres collagénases
3) Attachement (laminine et fibronectine)
4) Migration/motilité: (Fibronectine + chimioattraction auto par cyokines + paracrine par HGF et insuline like GF) |
|
|
qu'est ce que la migration amiboïde, avantage? |
la tumeur peut sauter la dégradation en faisant une migration amiboïde (les cellules «squeeze» à travers la matrice en utilisant les fibres de collagène comme voies). Ce processus se veut plus rapide. |
|
|
Qu'est-ce que l'anoïkose? comment l'éviter? |
forme spécifique d’apoptose, qui est due à un défaut d’interaction entre la cellule et la matrice extracellulaire.
Intéractions homotypiques (agrégation des cellules tumorales) Intéractions hétérotypiques (liaisons des cellules tumorales avec les plaquettes et leuko). |
|
|
Qu'est ce qui indique à la cellule tumorale de s'arrêter et de se fixer au tissu? |
- CAMs de la tumeur aux ligands endothéliaux -récepteur à chimiokines des sites - *nidation* des cellules tumorales par sécrétion de cytokines et facteurs de croissance.
C'est pourquoi on ne peut pas dire que ce n'est pas toujours au premier lit capillaire rencontré. (foie, poumons)
*exemple: Cd44R des tumeurs dans organes lymphoïdes via VEEL
|
|
|
Maladie relié au défaut de correction de l'ADN: - Mismatch - Excision de nucléotides - recombinaison homologue (dernier recours) |
Mismatch = HNPCC (cancer colon hereditaire non-polypeux. MSI instabilité + TGF-B, BAX
Excision= Xeroderma pigmentosum. U.V, cancer peau.
Recombinaison= -Radiation=syndrome de bloom, ataxia-telangiectasia -Nitrogen,DNA =anemie de fanconi - BRCA1 = ATM = détection dommage + p53 -BRCA2= RAD51 |
|
|
2 types d'antigènes tumoraux: |
-spécifiques: pas sur les cellules normales -associés: aussi sur les cellules normales
|
|
|
Types d'Ag tumoraux pouvant stimuler le système |
1. Gènes mutés 2. Protéines normales surexprimées 3. Ag de virus oncogéniques 4. Ag oncofétaux 5. Glycolipides/glycoprotéines de surface 6. Antigènes spécifiques de différenciation |
|
|
2 relais du système immunitaire pour éliminer cellules tumorales. |
NK: si pas de CMH1 vs CTLS: si CMH1
*Ne pas oublier macrophages avec TNF et ROS. *Ne pas oublier mécanisme humoral (anticorps) |
|
|
Mécanisme d'évitement du système immunitaire par les cellules tumorales. |
1. Croissance sélective des clones (élimination des immunogènes) 2. moins de CMH 3. Perte de costimulation (b7) 4. Immunosuppression : TGF-B , CTLa, 5. Masquage (glycocalyx) 6. Induction à l'apoptose des lymphocytes avec Fas-L sur cellules tumorales.
*rép immunitaire induit croissance tumorale par facteur croissance |
|
|
Qu'est ce que la signature métastasique? |
Prédisposition génétique à faire des métastases et qui serait le résultat d’anormalités multiples qui arriveraient plus tôt dans le développement de la tumeur.
Implication du stroma (micro-environnement) |
|
|
SNAIL et TWIST sont des candidats des oncogènes métastasiques qui déclancherait EMT.
Que veut-on dire par EMT |
Transition Epithéliale vers mésenchyme: -baisse de marqueurs épithéliaux et hausse de marqueurs dans mésenchyme qui contribue à un phénotype pro-migratoire des cellules tumorales |
|
|
Rôles ou contribution des cellules inflammatoires dans le cancer.
Macrophages: Fibroblastes: |
Macrophages: Formation et sécrétion de facteurs de métastases
Fibroblastes: réponse desmoplastique (remodelage du T.Conj) |
|
|
Explication de l'utilité de la glycolyse aérobique ou de l'effet Warburg. |
Oui, moins d'ATP ( 2 vs 38) et accumulation du pyruvate. Néanmoins, avantage de croissance dans un milieu hypoxique. Satisfaction de plusieurs cellules tumorales malgré la mince vascularisation.
Pyruvates accumulés utilisés: lipides, nucléotides pour faire face à leur prolifération cellulaire intense. |
|
|
V-F Pour faire face à leur environnement pauvre en nutriments, les cellules tumorales utilisent rapidement l'autophagie. |
Faux, à moins d'être dans des conditions très hostiles |
|
|
Changement chromosomique à l'origine du lymphome de Burkitt |
Translocation chromosome 8: Surexpression de MYC et IGH dans lymphocytes B. |
|
|
Changement chromosomique et implications du chromosome de Philadelphie |
chromosome 9 et 22 avec fusion des 2 gènes pour création d'une protéine chimérique BCR-ABL : activité tyrosine kinase continuelle (faux récepteur) |
|
|
2 pattern résultant de la surexpression de proto-oncogènes. |
Double minutes Régions homogènes stagnantes |
|
|
Sachant qu'un cancer altère ou amplifie l'expression de certain gène. Quels modifications épigénétiques sont sur les histones et l'ADN pour favoriser transcription? |
HyperAcétylation Déméthylation de l'ADN et des histones
*ou on peut mettre sous silence un gène suppresseur. |
|
|
Quels sont les 2 moyens que les miRNA ont de favoriser prolifération et cancer? |
oncogène: reduced miRNA, grande traduction, beaucoup d'oncoprotéine.
suppresseurs: abundant miRNA, pas de traduction, pas de protéine de répression.
Résultat commun: - Prolifération, évitement apoptose, capacité invasive, angiogénèse. |
|
|
Carcinogènes chimiques: |
1) Agents à action directe (pas d'activation): alkylants = électrphilie, liens covalents.
2) Agents à action indirecte: -Hydrocarbures polycycliques: Benzopyrene (cigarette) -Amines aromatiques: teinture d'Azo (cyp-450)
3) Naturels: Aflatoxine B1: Aspergillus (tp53)
4) Autres: Nitrite Biphenyls, Benzène/toluène, Chloryre de vinyle, amiante. |
|
|
Rôle des promoteurs dans la carcinogénèse chimique. (fumée, alcool) |
Suite à l'initiation par l'Agent carcinogène. Les Promoteurs peuvent induire tumeur ds ¢ initiées, mais pas directement (ne sont pas tumorogéniques par eux-mêmes). Dommages réversibles. Mais renforce la prolifération et le développement des mutations additionnelles menant à un néoplasme malin.
*carcinogène complet fait initiation et promotion. |
|
|
Par le rayonnement UV et la formation de dimères de pyrimidines, la réparation par excision de nucléotides peut être défaillante et faire place à quels cancers? |
Syndromes héréditaires rares: Xerodermapigmentosum,Bloom,Fanconi
o Carcinomes basocellulaires de la peau o Mélanomes malins |
|
|
Présentation cliniques des néoplasies. |
1) Leur localisation et leur impact sur les structures adjacentes (insuffisance endocrinienne, hypophysaire, obstruction intestinale)
2) Leurs fonctions (synthèse d’hormones et développement de syndromes paranéoplasiques)
3) Saignement et infections lorsque la tumeur ulcère les surfaces adjacents
4) Symptômes engendrés par des ruptures ou infarctus
5) Cachexie et dépérissement. (pas faim et lipolyse, Métabolisme augmenté, apport diminué par TNF, IL-1, LIF, PIF, LMF) |
|
|
Importance des syndromes paranéoplasiques - Cushing: corticostéroïdes - Hypercalcémie: PTHRP - neuromyopathies - Dermatologique: acanthosis nigricans - Ostéoarthropatie - Syndrome de Trousseau, DIC, Endocardite non Bactérienne
|
1) Peuvent constituer la manifestation la plus précoce d’une tumeur difficilement détectable.
2) Peuvent être cliniquement significatifs et provoquer la mort.
3) Peuvent confondre les traitements en imitant les symptômes d’une maladie métastatique. |
|
|
V-F. Le staging, basé sur l'histologie et l'observation des caractéristiques architecturales, est plus fiable cliniquement que le grading. |
faux, le staging est en fonction de grosseur lésion primaire, étendue de la propagation, et présence ou non de métastases. (TNM system)
Toutefois, en effet, plus efficace cliniquement que le grading. |
|
|
Exemple de virus à ARN carcinogène |
HTLV-1 et son influence sur leucémie cellule T. -TAX protein: NF-Kb = positive loop (cytokines, recepteur et costimulateurs) Il-15 - TAX inhibe p53 -Tax promote cyclines D -PROLIF polyclonale des TCD4, jusqu'à mutation additionnelle = monoclonale |
|
|
Mécanisme carcinogène du virus Epsteinn-Bar (EBV) sur lympho B. (pas de mort)
*lymphome burkitt (t(8,14))* |
CD21= LMP1 -vIL-10 : anti T-cell death - EBNa2: cyclin D - LMP1 vs MYC (pour échapper immunité) - promote BCL2 = prolif - par NFKB promote Jack/stat: Cd40L = prolif |
|
|
Mécanisme carcinogène du VPH |
E6: inhibe p53 E7: promote Rb et E2F et inhibe CDKinhib (p.21,27) |
|
|
V-F Le mécanisme carcinogène des virus hépatique B/C passe surtout par inflammation chronique |
vrai les cytokines, chemokines et GF stimule NF-KB qui inhibe apoptose. De plus, il y a expression de protéines HBx et HVc core pour une transduction accrue
|
|
|
V-F Le mécanisme carcinogène de la bactérie H.Pylori passe surtout par inflammation chronique |
Vrai, Longue évolution sur environ 10 ans: - Inflammation- atrophie - métaplasie - dysplasie- cancer (adénocarcinome)
ou
Lymphome B cells: - IL1 et tNF stimule TCD4 et B cells. - Quand lympho prolifère. Acquisition mutation et sont ensuite indépendant des T. - On parle alors d'un MALTomes. |
|
|
Facteurs anti-thrombotiques sécrétés par endothelium: |
- NO: dilat, inhib plaquette - PGI2: dilat, inhib plaquette -ADP phosphatase : fibrinogène + ADP -TFPI : inhibe complexe facteur 3,7,10. - t-PA: plasminogen activator -thrombomoduline + prot C + S = Va et VIIIa -antithrombine III + héparine = antithrombine |
|
|
Dessine moi la cascade de coagulation |
Bravo, |
|
|
Comment une agression cellulaire déclenche-t-elle l'inflammation aïgue? |
Extracellulaire (bacteria lipid ou polysaccharide) = TLR ou Lectine receptor = TNF + adhesion molecules
Intracellulaire (K+,ROS, Infection, ATP extra)= Inflammasome (CasP-1 + NLRP3) = IL-1
Dans les 2 cas, recrutement de neutrophiles, macrophage, Mastocytes , et cellules dendritiques.
|
|
|
Quelles sont les phases de guérison d'une plaie? |
1) Inflammatoire: caillot + cellules
2) Proliférative: 6 jours -tissue de granulation + angiogénèse+ collagène. 2e semaine- retrait angio, cicatrice blanche
3) Maturation: contraction et remodelage (TGFB+MMPs vs TIMPS) |
|
|
Récepteurs cellulaires importants pour la réparation tissulaire selon leur Growth factors.
*Transduction pathway*
|
-Kinase-Activity (HGF, EGF, VEGF, FGF)= RAS, PI3, PLC
- GCPR ( Chemokines, hormones )= GTP, AMPc , IP3, Ca++
- JAck/Stats à activité transcriptionnelle (cytokines, EPO) = direct au noyau comme F.Transcription |
|
|
Effets importants et multiples de la Kallikrenine, activée par le facteur Hagemann
Intégration entre inflammation et coagulation |
-Activation bradykinine (douleur, perméabilité) - Activation plasmine (complément C3 et C5, fibrine) - Activation directe complément (C5a, chimiotaxisme) |
|
|
Que sont les lymphos Treg? comment sont-ils différenciés? que font-ils? |
Pas d'infection...
TGF-B + IL-6 (comme Th17), mais un manque de Il-23 = Treg.
Treg inhibe Th1/Th2. |
|
|
4 sous-types de l'hypersensibilité de classe 4. (Cell mediated) |
IVa) Th1: IFN + Il-12 = Th1=TNF, IFN, Il-2 = macrophages = TNF, IL-1, Il-12
IVb) Th2 : éosino Il-4= Th2= Il4-5-13 = éosino + Mucus
IVc) CTL: citotoxicité CMH1= CD8 = CTLs= Perfo-granz-sergly + IFN-a vers IVa)
IVd) Th17: neutro Il-21, IL-23, Il-6, IL-1= Th17 = IL-17, Il-22, Il-21 = Neutrophiles
|
|
|
V-F Les changements post-mortem sont plus révélateurs que les marqueurs de la scène. |
faux. |
|
|
Caractéristiques des lividités post-mortem |
-Accumulation de sang dans petit vaisseaux selon les zones déclives du corps, sauf les points de pression
-8 à 12h à se fixer
-Si rupture, apparition de pétéchies
- Coloration peut être révélatrice: rouge cerise si CO. |
|
|
Caractéristiques des rigidités post-mortem |
-Complexe d'actine/myosine -Ce n'est pas une contraction, mais bien une raideur - À 20 celcius, 8-12h pour atteindre maximum, après disparition jusqu'à 36-48h - Si bris avant maximum, réapparition. - Si température ambiante chaude, plus de rigidités |
|
|
Caractéristiques des variations de température corporelle post-mortem. |
Rejoint température ambiante. -si à 20 celcius ambiant: 3h stable, suivi d'une descente de 1 degré/heure -Bcp de variables et facteurs peuvent influencer |
|
|
3 phénomènes caractérisant la décomposition. |
Autolyse: Enzymes intracellulaires détruisent tissus. Liquide ressemble à du sang. Exfoliation présente aussi (épiderme se sépare du derme également)
Putréfaction: Prolifération bactérienne, à partir tâche verte (24-36h). FID
Adipocire très lent phénomène (noyade): graisse (acide oléique. palmitique et stéarique deviennent de la cire, grisâtre)
*N.B que les vaisseaux peuvent se gonfler/distinguer par l'hémolyse+gaz |
|
|
Caractéristiques d'une blessure avec arme contondante. (poing, pied, barre, bâton) |
- Force, région, temps sont déterminants
- Érosion: Frottement, arrachement, pincement, impact
-Lacération: irrégulière, ponts tissulaires, décollement de la peau
-Hématome/contusion: Bleu/rouge vers violet/vert par accumulation d'hémosidérine (fer) et de bilirubine (hème).
|
|
|
Caractéristiques d'une blessure avec arme à bout piquant/tranchant: |
Piquant: plaie mince, mais profonde. (desfois plus que la lame, par compression
Tranchant: plaie plus longue que profonde
Érosion possible, présence de plaie de défenses. |
|
|
V-F Les machettes, haches ne peuvent pas vraiment être classées dans une catégorie d'arme. |
vrai. Ils sont techniquement coupants, mais la force de l'impact et leur grosseur les rendent contondants. |
|
|
Caratéristiques d'une plaie par balle
d'entrée:
Sortie: |
Entrée: définie, collerette érosive
Sortie: lacérations irrégulières
* direction du projectile peut aider à décrire l'agression * plusieurs indices quant à la proximité de tir |
|
|
Dans les os plats, quel est l'effet d'une plaie par balle. |
un trou en forme de cratère; au niveau du cratère d'entrée, le diamètre de l'orifice dans la table externe du crâne est inférieur à celui dans la table interne;
*c’est l’inverse pour la sortie* (plus petit interne, plus grand externe |
|
|
Indice(s) quant à la distance de tir. 1) bout touchant (touchant..) 2) très près (moins 1 pieds) 3) près (1 à 3 pieds) 4) loin (plus de 3 pieds) |
1) bout touchant (touchant..): - dommage des gaz, trou de mire, lacération en étoile, empreinte du canon
2) très près (moins 1 pieds): - noir de fumée (suie), tatouage de poudre, gaz (CO: rouge cerise des muscles touchés)
3) près (1 à 3 pieds) - tatouage de poudre
4) loin (plus de 3 pieds, et 5 pieds si long canon): - pas de poudre, mais dispersion des plombs peut aider. |
|
|
Parmi les AINS traditionnels, nommez les carboxyliques? |
Ac. Salicylique: AAS (aspirine) et salsalate
Proprionique: Ibuprophène (advil, motrin), Naproxène, Diclofenac (voltaren)
Indolacétique: indocid |
|
|
Parmi les AINS traditionnels, nommez le énoliques? |
Phenylbutazone |
|
|
Parmi les AINS traditionnels, nommez les oxycams? |
- Piroxicams (feldène) -Tenoxicam (Mobiflex) |
|
|
Pourquoi peut-il y avoir une atteinte digestive par l'utilisation des AINS. |
- Baisse le flot sanguin -Baisse HCO3- et mucus = Acidité
-Toxicité digestive d'origine SYSTÉMIQUE. (ulcère, perforation, hémorragie)
-Intolérance: dyspepsie, reflux, crampes |
|
|
6 facteurs à risques d'utilisation d'AINS |
1) +65 ans 2) ulcère depuis 10ans 3) Co-morbidité 4) Combinaison d'AINS (même si petite dose) 5) Anticoagulant 6) Prednisone (anti-inflammatoire glucocorticoïdes) |
|
|
Solutions possibles et fausses solution si facteurs à risque d'utilisation d'AINS |
Solution: 1) Éviter l'AINS 2) Cytoprotection avec un IPP (inhibiteur pompe protons) = Misoprostol, Omeprazol, pentoprozole
Fausses solutions: -Faible dose, courte période, Anti-H2, Cimétidine, Ranitidine |
|
|
V-F Les coxibs et glucocorticoïdes inhibe COx-1 |
FAux Cox-2 uniquement. Qui n'est pas systémique, mais bien inductible.
Tandis que AINS: inhibe les 2. |
|
|
En plus de l'effet anti-inflammatoire, quel effet majeur peut avoir un AINS |
Anti-Plaquettaire: empêche thrombose, mais très dangereux si déjà anti-coagulé sur coumadin ( ulcère, saignement, interférence avec coagulation) |
|
|
Pourquoi avantager les coxibs sur les AINS |
1) Sécurité digestive 2) Tolérance 3) Pas d'effet anti-plaquettaire
|
|
|
V-F Il y a un risque de toxicité rénale des AINS chez un patient avec condition pré-existante ou pré-rénale (cardiaque/ diurétique) |
vrai.
Risques de rétention hydro-sodée Hausse volume = HTA Protéinurie |
|
|
Bien qu'ils protègent la toxicité digestive, quel est le danger des coxibs |
-risque thrombique accru. -Accidents C-V -n'ont pas l'effet protecteur du naproxen
Pourquoi?: inhibition de PGI2 endothéliale (pas de Cox-1,ni COx-2) mais Cox-1 plaquettaire toujours actif avec production de Txa2 (aggrégation et vasoconstriction) |
|
|
Indication d'utilisation d'anti-inflammatoire si risque G-I avec arthrose, arthrite: |
1. Pas d'AINS 2. Coxibs+ AAS 3. AINS + IPP |
|
|
Indication d'utilisation d'anti-inflammatoire si risque C-V avec arthrose, arthrite: |
PAS D'AINS NI COXIBS |
|
|
Nommez les quatre étapes qui surviennent lors de la réparation tissulaire par la fibrose. |
1) Angiogénèse |
|
|
Caractéristiques d'une cellule tumorale maligne |
Macro: -Croissance accrue -Marges indéfinies -Métastases -Changements secondaires important. polymorphes
Micro: - faible différenciation : anaplasie - gros noyau hyperchromatique - ratio noyau/cyto: >>> - Mitoses importantes |
|
|
Phénomène de Tolérance centrale: Maturation des Lympho T dans thymus. 2 types de sélection? |
Positive: Sélection de ceux qui reconnaissent les CMH de soi
Négative: Élimination de ceux qui reconnaissent trop les antigènes de soi. |
|
|
Phénomène de la fièvre |
-LPS font sécréter IL-1 et TNF par leukocytes, plus de COX pour AA et PG.
-PGE2 relâchées dans cellules vasculaires et perivasculaires de l'hypothalamus.
-Ça met le setpoint de température corporelle plus élevé |
|
|
Quel type de leucocyte est particulièrement impliqué dans la lutte contre les cellules infectées ou tumorales? |
Lympho T cytotoxique |
|
|
Lequel n'est pas un AINS: -Piroxicam -Célécoxib -Cyclobenzaprine -Naproxen -Acide tiaprofénique |
Cyclobenzaprine |