Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
1231 Cards in this Set
- Front
- Back
|
Plummer-Vinson-Syndrom
|
Sideropenische Schleimhautatrophie von Zunge, Oropharynx und Ösophagus mit Zungenbrennen und schmerzhafter Dysphagie
|
|
|
Stadieneinteilung der Herzinsuffizienz
|
New York Heart Association (NYHA)
I Beschwerdefreiheit, normale körperliche Belastbarkeit II Beschwerden bei stärkerer körperlicher Belastung III Beschwerden schon bei leichter körperlicher Belastung IV Beschwerden in Ruhe Objektiv: I Belastbarkeit bis 150 Watt und mehr, VO2 > 25ml/kg/min II Belastbarkeit bis 100 Watt, VO2 15-25 ml/kg/min III Belastbarkeit bis 50 Watt, VO2 5-15 ml/kg/min IV Belastung nicht möglich, VO2 <5ml/kg/min |
|
|
Schilling Test
|
Vitamin B12 Resorptionstest
|
|
|
Klassifikation der pulmonalen Hypertonie
|
Gruppe I: Pulmonalarterielle Hypertonie (PAH), idiopathisch, familiär, assoziiert (z.B. mit Kollagenosen)
Gruppe II: Linksherzinsuffizienz Gruppe III: Lungenerkrankungen, Euler-Liljestrand-Reflex Gruppe IV: Chronische Thrombembolie |
|
|
Figlu Test
|
Nachweis des Folsäuremangels (orale Gabe von Histidin führt zu erhöhter Urinausscheidung von Formiminoglutamat)
|
|
|
Duke Kriterien für Endokarditis
|
Hauptkritierien:
1. Positive Blutkultur 2. Echokardiographischer Nachweis einer Vegetation, eines Abszess, einer neuen Klappeninsuffizienz Nebenkriterien: 1. prädisponierende Faktoren (Herzfahler, i.v. Drogenkonsum) 2. Temperaturerhöhung >= 38° 3. Vaskuläre Veränderungen (art. Embolisation, mykotisches Aneurysma, septische Lungeninfarkte, Janeway Läsion, konjunktivale Einblutung) 4. Immunologische Pathologien (Glomerulonephritis, Roth Spots, Osler Knötchen, pos. RF) 5. Mikrobielle Hinweise (pos. BK, die nicht den Hauptkriterien entspricht) Endokarditis nachgewiesen, wenn 2 HK, 1HK + 3 NK oder 5 NK Endokarditis möglich, wenn 1 HK + 1NK oder 3 NK |
|
|
Zieve-Syndrom
|
alkoholtoxischer Leberschaden, hämolytische Anämie, Hyperlipidämie
|
|
|
Positive Blutkultur als Hauptkritirium für Endokarditis
|
- Nachweis typischer Endokarditis-Erreger aus 2 getrennt abgenommenen Blutkulturen bei fehlendem anderen Fokus (Viridans Streptokokken, Streptococcus bovis, Staph. aureus, koagulasenegative Staphylokokken, Enterokokken, HACEK-Gruppe)
- persistierend positive Blutkulturen von positiven Endokarditis-Erregern (im Abstand von 12 Stunden) oder 3/3 bzw. 3/4 positiven Blutkulturen (Abstand 1 Stunde) von potentiellen Endokarditiserregern -Nachweis einer einzigen positiven Blutkultur von Coxiella burnetii oder IgG > 1:800 |
|
|
Gasser-Syndrom
|
Hämolytisch-urämisches Syndrom (HUS)
|
|
|
HACEK-Gruppe
|
gram-negative Bakterien, die eine Endokarditis verursachen können und schwierig nachzuweisen sind.
H aemophilus aphrophilus A ctinobacillus actinomycetemcomitans C Cardiobacterium hominis E ikenella corrodens K ingella kingae |
|
|
Moschcowitz-Syndrom
|
Thrombotisch-thrombozytopenische Purpura (TTP)
|
|
|
Endokarditis Libmann-Sacks
|
Manifestation bei Systemischem Lupus Erythematodes (SLE)
Abakterielle Endokarditis mit größeren Fibirinthromben auf der Mitral-, oder auf der Aorten- oder Pulmonalklappe. Häufig begleitet von Perikarditis und Pleuritis. |
|
|
Heinz-Innenkörperchen
|
Hämoglobin-Präzipitate, z.B. bei G-6-PD-Mangel
|
|
|
SIRS
|
Systemic Inflammatory Response Syndrome:
Generalisierte inflammatorische Reaktion unterschiedlicher Genese. >= 2 Kritierien: 1. Temperatur > 38° oder <36° 2. Tachykardie > 90/min 3. Tachypnoe > 20/min 4. pCO2 <32mmHg 5. Leukozyten > 12.000/yl oder < 3800/yl; stabkernige/ unreife Neutrophile > 10% |
|
|
Howell-Jolly-Körperchen
|
Kernreste in Erythrozyten bei fehlender Milz
|
|
|
Septischer Schock
|
3 Diagnosekriterien:
1. Nachweis einer Bakteriämie (positive Blutkultur) 2. SIRS 3. Art. Hypotension trotz ausreichender Volumensubstitution (RR sys < 90mmHg oder MAP <70mmHg |
|
|
Cooley-Anämie
|
Majorform der Beta-Thalassämie
|
|
|
Respiratorische Azidose
|
Keine Indikation für die Gabe von Bicarbonat.
Bicarbonat kummuliert im Knochen, Muskel und Fettgewebe, weswegen es manchmal Tage dauern kann, bis deutlich erhöhte pCO2 Werte auch unter suffizienter Beatmung sinken, da es immer wieder vom gespeicherten Bicarbonat nachgeliefert wird. |
|
|
Marchiafava Anämie
|
Paroxysmale nächtliche Hämoglobinurie
|
|
|
Anionenlücke
|
Natrium - (Chlorid + Bicarbonat)
Eingrenzung der Ursache einer metabolischen Azidose Normwert: 12 +- 4mmol/l Vergrößerte Anionenlücke: Der Bicarbonatverbrauch wird durch nicht-gemessene Anionen ausgeglichen, zB. Urämie, Ketoazidose, Laktatazidose, Gifte Normale Anionenlücke: Der Verlust an Bicarbonat wird durch Chlorid ausgeglichen (hyperchlorämische Azidose, Substraktionsazidose), z.B. renal tubuläre Azidose, Diarrhoe |
|
|
Budd-Chiari-Syndrom
|
Verschluss der Lebervenen durch Thrombosen oder Tumor
|
|
|
Autoregulation der Niere
|
Bayliss-Effekt
Für die Filtration ist ein konstanter Blutdruck notwendig. Um den Perfusionsdruck konstant zu halten, hat die Niere die Fähigkeit zur Autoregulation (Barorezeptoren im Vas afferens und Vas efferens) |
|
|
Cruveilhier-von-Baumgarten-Syndrom
|
Venöse Verbindung zwischen Umbilikalvenen und epigastrischen Venen
|
|
|
ALI
|
Acute Lung Injury
-Akuter Beginn -PO2/ FIO2 <= 300mmHg -Bilaterale Infiltrate -PCWP <= 18mmHg |
|
|
Eculizumab (Soliris)
|
Anti-C5-mAB
Spezifische Therapie bei PNH (paroxysmale nächtliche Hämoglobinurie) Meningokokken-Impfung! |
|
|
ARDS
|
Acute Respiratory Distress Syndrome
-Akuter Beginn -PO2/ FIO2 <= 200mmHg -Bilaterale Infiltrate -PCWP <= 18mmHg Schweregradeinteilung nach dem Lung Injury Score nach Murray (Röntgenbefund, PO2/FIO2, PEEP, Compliance) Generalisierte pulmonale Entzündungsreaktion mit konsekutiver Permeabilitätsstörung (Capillary leak syndrome), die zu einem nicht kardiogenen Lungenödem führt (Austritt von Plasmaproteinen ins Interstitium und in die Alveolen) |
|
|
Direkter und indirekter Coombs-Test
|
Direkter Coombs-Test: Nachweis von igG-AK, die an Erythrozyten haften
Indirekter Coombs-Test: Nachweis von IgG-AK gegen Erythrozyten, die noch frei im zu untersuchenden Serum vorhanden sind (z.B. Nachweis von inkompletten IgG-AK im Serum der Mutter bei Rh-Inkompatibilität) |
|
|
Ätiologie ARDS
|
Pulmonales ARDS:
-Pneumonie -Aspiration, Beinahe-Ertrinken -Inhalationstrauma (z.B. Rauchgas) -Thoraxkontusion -Lungenembolie Extrapulmonales ARDS -Sepsis -Peritonitis -Pankreatitis -Polytrauma -Verbrennungskrankheit -Herz-Lungen-Maschine -DIC -Leber-/ Nierenversagen -Eklampsie |
|
|
Donath-Landsteiner-Test
|
Bithermische Hämolysine binden sich bei kalten Temperaturen mit Komplement an Erythrozyten und führen bei Erwärmung (37°C) zu Hämolyse.
|
|
|
Pathophysiologie ARDS
|
-Mikrothrombosen in der pulmonalen Strombahn
-Bildung und Freisetzung endogener vasokonstriktiv wirkender Mediatoren (Thromboxan, Endothelin) -Hypoxische pulmonale Vasokonstriktion |
|
|
Café au lait -Farbe der Haut
|
Renale Anämie (anämische Blässe und Ablagerung von Urochromen)
|
|
|
Stadien ARDS
|
1. Exsudatives Stadium: Capillary leak -> interstitielles Lungenödem
2. Untergang der Typ II Pneumozyten -> verminderte Bildung von Surfactant -> alveoläres Lungenödem 3. Proliferative Phase: Lungenfibrose und Endothelproliferation der Alveolarkapillaren |
|
|
Fanconi Anämie
|
angeborene aplastische Anämie
|
|
|
Ramsay Score
|
R1: Pat. wach, agitiert, unruhig, ängstlich
R2: Pat. auf akkustische Reize weckbar, ruhig, kooperativ, orientiert, Beatmungstoleranz R3: Pat. schläft, lebhafte Reaktion auf manuellen Stimulus R4: Pat. schläft, träge Reaktion auf manuellen Stimulus oder lautes Geräusch R5: Pat. schläft, Reaktion nur auf starke Schmerzreize R6: Pat. schläft, keinerlei Reaktion auslösbar |
|
|
Morbus Werlhof
|
Immunthrombozytopenische Purpura (ITP)
|
|
|
Howell-Jolly-Körperchen
|
Kernreste in Erythrozyten bei fehlender Milz
|
|
|
Waterhouse-Friderichsen-Syndrom
|
Fulminante Meningokokkensepsis mit Verbrauchskoagulopathie, NNR-Blutungen)
|
|
|
Pathophysiologsiche Einteilung der Anämien
|
1. Bildungsstörungen
2. Gesteigerter Abbau (korpuskulär oder extrakorpuskulär) 3. Verlust (Blutungen) 4. Verteilungsstörungen (Hyperspelnie) |
|
|
Rumpel-Leede-Test
|
Nach 5 Minunten venöser Stauung (20mmHg unter syst. RR) treten im positiven Fall punktförmige Blutungen am Unterarm auf
Angiopathien, Thrombopenien, Thrombozytopathien |
|
|
Laborparameter bei der Eisenmangelanämie
|
-löslicher Transferrinrezeptor erhöht (unbeeinflusst von Entzündungszuständen)
-Ferritin erniedrigt -Serumeisen erniedrigt -Transferrinsättigung erniedrigt Ein prälatenter Eisenmangel ist lange vor Erschöpfung der Eisenspeicher an einer verminderten Ferritinkonzentration und einem Anstieg des sTfR erkennbar. |
|
|
OPSI
|
Overwhelming postsplenectomy infection: Akute Pneumokokkensepsis mit DIC und hoher Letalität
|
|
|
Differentialdiagnose der hypochromen Anämie
|
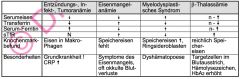
|
|
|
von-Willebrand-Jürgens-Syndrom
|
vWF und F. VIIIC vermindert
Patienten neigen weniger zu spontanen Blutungen als bei Hämophilie. |
|
|
Plummer-Vinson-Syndrom
|
Sideropenische Schleimhautatrophie von Zunge, Oropharynx, Ösophagus mit Zungenbrennen und schmerzhafter Dysphagie
|
|
|
CRB-65-Index
|
Sterblichkeit der CAP, ab 1-2 Punkten stationäre Aufnahme
Confusion Respiratory Rate >30/min Blood Pressure (<90mmHg systolisch) Alter über 65 Jahre |
|
|
Symptome des Eisenmangels
|
1. Haut/ Schleimhäute: Rillenbildung der Nägel, Hohlnägel (Koilonychie), diffuser Haarausfall, Aphten, trockene Haut, Pruritus, Plummer-Vinson-Syndrom
2. Neurologisch: Kopfschmerzen, Konzentrationsstörungen, leichte Erregbarkeit, restless-legs, Pica 3. Anämie: Blässe, Schwäche, Belastungsdyspnoe, Systolikum, Tachykardie |
|
|
Osler Knötchen
|
kleine subkutane, schmerzhafte, entzündlich gerötete, hämorrhagische Effloreszenzen, die meist Zeichen einer Mikroembolie oder Immunkomplexvaskulitis im Rahmen einer infektiösen Endokarditis sind. Sie treten insbesondere an Finger- und Zehenkuppen, sowie im Thenar- und Hypothenargebiet auf, aber auch an Armen und Beinen. Charakteristisch ist eine gruppenförmige Anordnung der oft zu Hunderten auftretenden Knoten. Sie heilen meist nach wenigen Tagen unter Schuppung aus.
|
|
|
Eisenmangelanämie Blutbildveränderungen
|
mikrozytär (MCV < 80fl)
hypochrom (MCH < 28pg) Poikilozytose Anisozytose |
|
|
Janeway Lesions
|
Janeway-Läsionen sind kleine erythematöse oder hämorrhagische Flecken oder Knoten der Handinnenflächen oder Fußsohlen.
Sie sind pathognomonisch für infektiöse (bakterielle) Endokarditis. Ihre Pathogenese beruht auf einer Typ III Hypersensitivitätsreaktion. |
|
|
Wie lange reichen Vit B12 und Folsäure bei fehlender Zufuhr?
|
Vit B12: 3 Jahre
Folsäure: 3 Monate |
|
|
Milchglastrübungen
|
im CT Zeichen einer Alveolitis -> BAL
|
|
|
Trias bei schwerem Vit B12 Mangel
|
1. Anämie mit Café au lait Farbe der Haut (diskreter Ikterus durch ineffektive Erythropoese)
2. Gastrointestinale Symptome: atrophische Glossitis (Hunter-Glossitis), ggf. Autoimmungastritis Typ A 3. Neurologische Symptome: Funikuläre Meylose (Hinterstränge und Pyramidenbahn; Stimmgabelversuch) |
|
|
Hamman-Rich-Syndrom
|
Akute interstitielle Pneumonie
|
|
|
Morbus Biermer
|
Perniziöse Anämie
Auto-Antikörper gegen Parietalzellen und Intrinsic factor mit atrophischer Autoimmungastritis vom Typ A und Achlorhydrie |
|
|
Morbus Boeck
|
Sarkoidose
|
|
|
Hunter Glossitis
|
atrophische Glossitis mit glatter roter Zunge und Zungenbrennen bei Vit B12 Mangel
|
|
|
Löfgren-Syndrom
|
Akute Verlaufsform der Sarkoidose:
-Erythema nodosum -bihiläre Lymphadenopathie -Polyarthritis (Sprunggelenksarthritis) -Allgemeinsymptome |
|
|
Ursachen für Vit B12 Mangel
|
1. streng vegetarische Kost
2. Mangel an intrinsic factor (Z.n. Magenresektion, perniziöse Anämie) 3. Malabsorption 4. Fischbandwurm (selten manifest) 5. blind loop Syndrom mit bakterieller Überwucherung |
|
|
Heerfordt-Syndrom
|
Parotitis, Uveitis, Fascialisparese bei Sarkoidose
|
|
|
Ursache der megalobalstären Anämie beim Alkoholiker
|
Folsäure Mangel
|
|
|
Morbus Jüngling
|
Knochenbefall der Sarkoidose im Bereich der Endphalangen
|
|
|
Resorptionstests bei megaloblastärer Anämie
|
1. Schilling Test: Vit B12 Resorptionstest (mit und ohne IF)
2. Figlu Test: orale Gabe von Histidin -> erhöhte Ausscheidung von Formiminoglutamat bei Folsäuremangel |
|
|
CREST-Syndrom
|
Calcinosis cutis
Raynaud-Syndrom Ösophagusbeteiligung Sklerodaktylie Teleangiektasien häufig pulmonale Hypertonie |
|
|
Vit B12 Substitution
|
initial 1000yg/ Woche i.m. bis zur Normalisierung des Blutbildes, danach lebenslang 1000yg alle 3 bis 6 Monate.
Zu Beginn der Therapie Kalium und Eisen nach Werten substituieren! (Cave Herzrhyhtmusstörungen) |
|
|
Sjögren-Syndrom
|
Lymphozytäre Infiltration von Tränen- und Speicheldrüsen
dry eye, dry mouth erhöhte Lymphominzidenz sekundär bei Lymphomen |
|
|
Normale Überlebenszeit von Erythrozyten
|
120 Tage
|
|
|
Schirmer-Test
|
Nachweis einer verminderten Tränensekretion (Filterpapierstreifen über Unterlid)
|
|
|
Korpuskuläre hämolytische Anämien
|
1. angeborene Membrandefekte (z.B. Sphärozytose)
2. angeborene Enzymdefekte (z.B. G-6-PD-Mangel, PK-Mangel) 3. Hämoglobinopathien (Thalassämie) 4. erworbene Membrandefekte (paroxysmale nächtliche Hämoglobinurie) |
|
|
Saxon-Test
|
Messung der Speichelproduktion (Abwiegen eins Wattebausches, der 2 Minuten in den Mund genommen wurde)
|
|
|
Favismus
|
Glukose-6-Phophatdehydrogenase-Mangel (G-6-PD-Mangel), führt zu verminderter Bildung von reduziertem Glutathion, welches die Erythrozyten vor Oxidationsschäden schützt.
Häufig bei Afrikanern, Asiaten und Bewohnern des Mittelmeerraumes, Vererbung x-chromosomal rezessiv. Auslösung hämolytischer Krisen durch Infektionen, Genuss von Saubohnen, Chinin, Primaquin, Chloroquin, Sulfonamide, ASS) Typisch: Heinz´Innenkörperchen (Denaturierungsprodukte des Hämoglobin) Meidung auslösender Noxen, Patientenausweis. |
|
|
Anti-Jo 1-Syndrom
|
Myositis
Raynaud-Syndrom Arthritis Lungenfibrose |
|
|
Medikamentöse Therapieindikation für Hyperurikämie
|
Harnsäure > 9mg/dl oder Gichtanfälle. Bis 9mg/dl nur diätetische Behandlung.
Cave Allopurinol-Hypersensitivitätssyndrom (Vaskulitis mit Dermatitis, Hepatitis und Nierenversagen) |
|
|
Lilakrankheit
|
Dermatomysitis
|
|
|
Nebenwirkungen und Wechselwirkungen von Allopurinol
|
Leukopenie, GI-Symptome.
Gefährliche NW: Allopurinol-Hypersensitivitätssyndrom (Vaskulitis mit Dermatitis, Hepatitis und Nierenversagen, Eosinophilie im Blut) WW: Hemmung des Abbaus von Azathioprin (Dosisreduktion um 75%), Theophyllin und Phenprocuomon Cave: Therapiebeginn nicht im akuten Gichtanfall, da die Auflösung von Uratkristallen Gichtanfälle begünstigen kann. |
|
|
Gottron Papeln
|
lichenoide weißliche Papeln an den Fingerstreckseiten bei Polymyositis/ Dermatomyositis
|
|
|
Auslöser von Gichtanfällen
|
Fasten und Feste
|
|
|
Keinig-Zeichen
|
druckschmerzhafte Nagelfalzhyperkeratosen bei Polymyositis/ Dermatomyositis
|
|
|
Therapie im akuten Gichtanfall
|
1. Wahl: NSAR, ggf. Prednisolon 20mg
2. Wahl: Colchicin, wegen NW Reservemittel |
|
|
Chvostek Zeichen
|
Beim Beklopfen des Nervus facialis im Bereich der Wange wird im positiven Fall Zucken der Mundwinkel ausgelöst
Zeichen der Hypokalzämie |
|
|
Einteilung der hämolytischen Anämien
|
I Korpuskuläre hämolytische Anämien
1. Angeborene Membrandefekte (z.B. Sphärozytose, Elliptozytose) 2. Angeborene Enzymdefekte (z.B. G-6-PD-Mangel, PK-Mangel) 3. Angeborene Hämoglobinopathien (z.B. Thalassämie) 4. Erworbene Membrandefekte (z.B. paroxysmale nächtliche Hämoglobinurie) II Extrakorpuskuläre hämolytische Anämien 1. Isoantikörper (z.B. Transfusionsreaktion) 2. AIHA 3. Malaria (infektiös) 4. Herzklappenersatz (mechanisch) 5. Zieve-Syndrom (Stoffwechsel) 6. Mikroangiopathisch (HUS, TTP) |
|
|
Trousseau Zeichen
|
Blutdruckmanschette -> Pfötchenstellung
Zeichen der Hypokalzämie |
|
|
Laborparameter bei intravasaler Hämolyse
|
-niedriges (freies) Haptoglobin, später niedriges Hämopexin
-LDH-Erhöhung -erhöhtes indirektes Bilirubin -erhöhte Retikulozyten -freies Hämoglobin im Serum -Hämoglobinurie -Im Urin Bilirubin negativ, Urobilinogen pos. |
|
|
Jacksonian Democracy
|
The Jacksonian era (1829-1841) included many reforms: free public schools, more women's rights, better working conditions in factories, and the rise of the Abolition movement. In the election, Jackson was portrayed as a common man and his opponent, J.Q. Adams, was attacked for his aristocratic 'principles.' Electors in the Electoral College were also chosen by popular vote. [Common man, nationalism, National Nominating Conventions.]
|
|
|
Bilirubin
|
indirekt: Hämolyse
direkt: Cholestase |
|
|
Missouri Compromise
|
Admitted Missouri as a slave state and at the same time admitted Maine as a free state. Declared that all territory north of the 36 30 latitude would be prohibited to slavery. Was declared unconstitutional by Dred Scott vs Sanford.
|
|
|
Symptome der akuten hämolytischen Krise
|
-Fieber, Schüttelfrost
-Hb-Abfall, Ikterus, Hyperbilirubinämie -Kopf-, Bauch- und Rückschmerzen -bierbrauner Urin (Hämoglobinurie) |
|
|
Goddpasture Syndrom
|
Antibasalmembran RPGN mit Lungenbeteiligung (Hämoptysen)
Junge Männer, selten, Immunsuppressive Therapie, Plasmapherese |
|
|
G-6-PD-Mangel
|
Glucose-6-Phosphatdehydrogenase-Mangel:
-autosomal rezessiver Erbgang -verminderte Bildung von reduziertem Glutathion, welches die Erythrozyten vor Oxidationsschäden schützt -Auslösung hämolytischer Krisen durch oxidativen Stress: Infektionen, Saubohnen (Fava), Chloroquin, Chinin, ASS, Sulfonamide) -typisch: Heinz-Innenkörperchen (Denaturierungsprodukte des Hb) -Patientenausweis, Meidung auslösender Noxen -Häufig bei Afrikanern, Asiaten und Bewohnern des Mittelmeerraumes |
|
|
Diffuse alveoläre Hämorrhagie
|
DAH
Kapillaritis Trias: erhöhte CO-Diffusionskapazität, schwere Hypoxämie, diffuse Lungeninfiltrate Ät.: Kollagenosen, Vaskulitiden, Medikamente, ARDS, Lungeninfarkt |
|
|
Sichelzellanämie
|
-autosomal kodominant: Punktmutation im Beta-Globinlokus auf Chromosom 11 führt zur Produktion von HbS, das im desoxigenierten Zustand präzipitiert (Sichelform) (80% HbS, 20% HbF)
-Vasookllusion, Organinfarkte -gesteigerte Neigung zu bakteriellen Infekten (Atrophie der Milz) -aseptische Knochennekrosen -Niereninsuffizienz -Heterozygote Fälle meist beschwerdefrei, homozygote oft schwerer Verlauf -Sichelzelltest: Tropfen Blut, Deckglas luftdicht, nach 24 Stunden Sichelzellen |
|
|
IgA-Nephropathie
|
Morbus Berger
Immunkomplexablagerung von IgA im Glomerulum idiopathisch oder sekundär (Purpura Schoenlein Hennoch, SLE, RA, Leberzirrhose, Sprue, Psoriasis, Dermatitis herpetiformis During) 1-3 Tage nach unspez. Infekt der oberen Luftwege Makrohämaturie Behandlung nach Höhe der Proteinurie (abwarten, ACE-Hemmer, Kortikoide) |
|
|
Erkrankungen mit partieller Resistenz gegen Malaria
|
-Sichelzellenanämie
-G-6-PD-Mangel -Thalassämie |
|
|
Häufige Ursachen für chronische Niereninsuffizienz
|
1. Diabetische Nephropathie (35%)
2. Glomerulonephritis (15%) 3. Chronisch tubulo-interstitielle Erkrankungen 4. hypertensive Nephropathien 5. Polyzystische Nierenerkrankungen |
|
|
Beta-Thalassämie
|
Minorform (Heterozygotie), Majorform (Cooley-Anämie, Homozygotie)
-verminderte Produktion von Beta-Ketten -> HbF/ HbA2 erhöht durch vermehrte Produktion von delta- oder gamma-Ketten -Targetzellen, basophile Tüpfelung -Bei Majorform von Kindheit an Dauertherapie mit Transfusionen und Eisenelimination notwendig, ggf. allogene Knochenmarkstransplantation |
|
|
Akute postinfektiöse Glomerulonephritis
|
Immunkomplexnephritis nach Infekt mit Beta-hämolysierenden Streptokokken
Beschwerdefreies Intervall von 1-2 Wochen Volhard´ Trias: Hämaturie, Hypertonie, Ödeme Erythrozytenzylinder Kinder > 90% Ausheilung Erwachsene nur 50% Ausheilung chronische Verlaufsformen möglich Therapie: körperliche Schonung, Sanierung des Streptokokkeninfektes, Behandlung von Komplikationen |
|
|
Paroxysmale nächtliche Hämoglobinurie
|
Marchiafava-Anämie
Erworbene klonale Erkrankung der pluripotenten hämatopetischen Stammzelle mit Störung des GPI (Glykosyl-Phophatidyl-Inositol)-Ankers. Unzureichender Schutz der Erythrozytenmembran, Komplementaktivierung durch Infekte oder Abfall des Blut pH Klinik: Trias aus Hämolyse, Thrombosen und Zytopenie (Thrombosen durch Aktivierung der Thrombozyten durch NO-Depletion und Kontakt mit innerer Erythrozytenmembran) Komplikationen: Übergang in MDS oder AML, chronische Niereninsuffizienz, pulmonale Hypertonie Therapie: Allogene Stammzelltransplantation, Eculizumab (Anti C5-mAB), supportive Therapie |
|
|
Volhard´Trias
|
bei akuter postinfektiöser Glomerulonephritis:
Hämaturie Hypertonie Ödeme |
|
|
Transfusionszwischenfall
|
1. Hämolytische Transfusionsreaktion
2. Allergische Transfusionsreaktion durch AK gegen HLA-AG der Leukozyten oder Plasmabestandteile 3. Septische Reaktionen durch Kontamination der Konserve 4. Fieberhafte Reaktion durch Pyrogene in der Konserve 5. Transfusionsassoziierte Lungeninsuffizienz (TRALI) |
|
|
Morbus Berger
|
IgA-Nephropathie (idiopathische Immunkomplex-GN mit Ablagerung von IgA im Glomerulum)
|
|
|
Verhalten bei Transfusionszwischenfall
|
-Transfusion stoppen
-Volumen -NaBi zur Prophylaxe eines akuten Nierenversagens -antiallergische Therapie -Thrombembolieprophylaxe |
|
|
Dermatitis Herpetiformis Duhring
|
Hautmanifestation bei glutensensitiver Enteropathie
Erytheme, Plaques, herpetiforme Böäschen, besonders gluteal, am Schultergürtel und an den Streckseiten der Extremitäten Juckreiz! |
|
|
AIHA
|
Autoimmunhämolytische Anämien
1. AIHA durch inkomplette Wärmeautoantikörper IgG -> extravasale Hämolyse (Milz, RES); hohe BSG, Schwierigkeiten beim Ablesen der Kreuzprobe; direkter Coombs-Test positiv; Vorkommen: idiopathisch, NHL, M. Hodgkin, SLE, Medikamente, Virusinfekte 2. AIHA durch Kälteagglutinine IgM ->intravasale Hämolyse; passager meist 2-3 Wochen nach Infekt (Mykoplasmen) oder chronisch (idiopathisch oder bei B-Zell-Lymphomen, Hep. C); hohe BSG, Schwierigkeiten bei der Blutabnahme; Leitsymptom: Akrozyanose 3. AIHA durch bithermische Hämolysine; Donath-Landsteiner-Typ; passager bei Infekt Therapie: Wärmeauto-AK: Kortikoide, Immunsuppresiva, Splenektomie, hoch dosierte Gabe von Immunglobulinen bei hämolyt. Krise Kälteauto-AK: Schutz vor Kälte, Kortikoide und Splenektomie sind unwirksam (intravasale Hämolyse), evtl. Plasmapherese Beide: Supplementierung von Folsäure und Vit B12 |
|
|
Waschmaschinen-Phänomen
|
Dünndarmschlingen mit vermehrter Flüssigkeitsfüllung und verdickter Wand, vor- und rückwärts gerichtete Peristaltik
Vorkommen: Sprue |
|
|
Mononukleose-ähnliches Krankheitsbild
|
EBV
CMV HIV Fieber, LK-Schwellungen, Splenomegalie, Angina, Myalgien |
|
|
Marsh-Kriterien
|
Dünndarmbiopsie bei glutensensitiver Enteropahtie
Zottenatrophie, Kryptenhyperplasie |
|
|
Normale Lebensdauer von Thrombozyten
|
9-10 Tage
|
|
|
ESPGAN-Kritierien
|
European Society for Paediatric Gastroenterology and Nutrition 1990: Histologie und Besserung nach glutenfreier Diät zur Diagnose einer Sprue
|
|
|
HUS
|
Hämolytisch urämisches Syndrom (Gasser-Syndrom)
-gehört wie die TTP zu den thrombotischen Mikroangiopathien -typisch bei Kindern nach EHEC-Infekt -atypisches HUS als familiäre und sporadische Form -Endothelschwellung und Ablösung -> mikroangiopathische hämolytische Anämie, Niereninsuffizienz und akute Thrombozytopenie -Frischplasma/ Plasmapherese |
|
|
Cronkhite Canada Syndrom
|
Generalisierte Polypose des GIT und bräunliche Hautpigmentierung mit Alopezie und Nagelveränderungen, manifestiert sich nach dem 50 LJ, therapierefraktäre Diarrhoe, KRK-Risiko nicht erhöht
|
|
|
TTP
|
Thrombotisch thrombozytopenische Purpura (Moschcowitz-Syndrom)
-angborener Mangel an vWF-cleaving-Protease oder erworbene AK-Bildung gegen vWF-cleaving-Protease gehört wie die HUS zu den thrombotischen Mikroangiopathien -Endothelschwellung und Ablösung -> mikroangiopathische hämolytische Anämie, Niereninsuffizienz und akute Thrombozytopenie -Frischplasma/ Plasmapherese |
|
|
Peutz-Jeghers-Syndrom
|
Polypose des Dünndarms, gel. auch des Dickdarms, 50% autosomal dominant, 50% Neumutationen
Melaninflecken an Lippen und Mundschleimhaut gel. Stieldrehung der Polypen mit hämorrhagischer Infarzierung erhöhtes Risiko für Pankreas-Karzinom, KRK und Ovarial Karzinom |
|
|
ITP
|
Immunthrombozytoppenische Purpura (M. Werlhof)
-isolierte Thrombozytopenie ohne erkennbare Ursache mit verkürzter Plättchenüberlebenszeit; Autoimmunpathogenese (AK gegen GpIIb/ IIIa) -Helicobacter pylori bei einem Teil der Patienten pathogenetische Rolle -> Eradikation -Kortikoide, H.p.-Eradikation, Immunglobuline präoperativ und präpartal, evtl Rituximab, Rombiplostim (Thrombopoese stimulierendes Medikament); Splenektomie -Thrombozytensubstitution nur bei schweren Blutungen |
|
|
Cronkhite Canada Syndrom
|
Generalisierte Polypose des GIT und bräunliche Hautpigmentierung mit Alopezie und Nagelveränderungen, manifestiert sich nach dem 50 LJ, therapierefraktäre Diarrhoe, KRK-Risiko nicht erhöht
|
|
|
HIT
|
Heparininduzierte Thrombozytopenie
2 Formen: HIT I: in den ersten Tagen der Behandlung mit UFH Thrombozytenabfall < 30% des Ausgangswertes, spontane Normalisierung, weitere Heparingabe möglich HIT II: AK- Bildung, bei nicht sensibilisierten Patienten zwischen dem 5. und 14. Tag; White Clot Syndrom: Thrombosen in 25% der Fälle! HIT II bei NMH 30x seltener als bei UFH |
|
|
Cowden Syndrom
|
Papeln im Gesicht, an Händen, Füßen, Papillome im Mundbereich, harmatöse Tumoren (Mamma, Schilddrüse) und Polypen im GIT
Erhöhtes Risiko für KRK |
|
|
Therapie DIC
|
Prä-DIC-Phase: Heparin
Manifestes DIC: FFP, AT-III, ggf. Thrombozytenkonzentrate, kein Heparin! Post-DIC-Phase: Heparin |
|
|
Peutz-Jeghers-Syndrom
|
Polypose des Dünndarms, gel. auch des Dickdarms, 50% autosomal dominant, 50% Neumutationen
Melaninflecken an Lippen und Mundschleimhaut gel. Stieldrehung der Polypen mit hämorrhagischer Infarzierung erhöhtes Risiko für Pankreas-Karzinom, KRK und Ovarial Karzinom |
|
|
Impfungen vor geplanter Splenektomie
|
Penumokokken, Haemophilus influenzae Typ b (Hib), Meningokokken
|
|
|
Cowden Syndrom
|
Papeln im Gesicht, an Händen, Füßen, Papillome im Mundbereich, harmatöse Tumoren (Mamma, Schilddrüse) und Polypen im GIT
Erhöhtes Risiko für KRK |
|
|
Hämorrhagische Diathesen
|
1. Thrombozytopnie, Thrombozytopathie (70%)
2. Plasmatisch (20%) 3. Vaskulär (10%) |
|
|
Cronkhite Canada Syndrom
|
Generalisierte Polypose des GIT und bräunliche Hautpigmentierung mit Alopezie und Nagelveränderungen, manifestiert sich nach dem 50 LJ, therapierefraktäre Diarrhoe, KRK-Risiko nicht erhöht
|
|
|
Hämophilie
|
Typ A: Fehlen oder Inaktivität von Faktor VIII
Typ B: Fehlen oder Inaktivität von Faktor IX (Christmas Faktor) X-chromosomal rezessiv Großflächige Blutungen, Muskelblutungen, Gelenkblutungen PPT verlängert, INR normal |
|
|
Peutz-Jeghers-Syndrom
|
Polypose des Dünndarms, gel. auch des Dickdarms, 50% autosomal dominant, 50% Neumutationen
Melaninflecken an Lippen und Mundschleimhaut gel. Stieldrehung der Polypen mit hämorrhagischer Infarzierung erhöhtes Risiko für Pankreas-Karzinom, KRK und Ovarial Karzinom |
|
|
ambulante Behandlung der CAP
|
Risikofaktoren: AB-Vortherapie in den letzten 3 Monaten, Bewohner von Pflegeheimen, chronische Begleiterkrankungen
Ohne Risikofaktoren: Amoxicillin, Alternativen: Makrolide, Doxycyclin Mit Risikofaktoren: Amoxicillin/ Clavulansäure (Augmentan) Alternativen: Levofloxacin, Cephalosporin |
|
|
Cowden Syndrom
|
Papeln im Gesicht, an Händen, Füßen, Papillome im Mundbereich, harmatöse Tumoren (Mamma, Schilddrüse) und Polypen im GIT
Erhöhtes Risiko für KRK |
|
|
stationäre Behandlung der CAP
|
Risikofaktoren für P. aeruginosa: Pulmonale Komorbidität, Kortikoide > 4 Wochen, AB-Vorbehandlung, Krankenhausaufenthalt in den letzten 30 Tagen
Ohne Risikofaktoren für Pseudomonas: Augmentan, evtl. + Makrolid Cefuroxim/ Ceftriaxon evtl. + Makrolid oder Levofloxacin Mit Risikofaktoren für Pseudomonas: Tazobac + Levo- oder Ciprofloxacin Zienam + Levo- oder Ciprofloxacin Fortum (Ceftazidim) + Levo- oder Ciprofloxacin |
|
|
AWMF
|
Arbeitsgemeinschaft der Wissenschaftlichen medizinischen Fachgesellschaften
|
|
|
Behandlung der nosokomialen Pneumonie
|
Risikofaktoren:
Alter > 65 Jahre (1 Punkt) Strukturelle Lungenerkrankung (2) AB-Vorbehandlung (2) Late Onset (ab 5. KH-Tag) (3) Schwere resp. Insuff. (3) Extrapulm. Organversagen (4) Je mehr Punkte, umso breitere Behandlung |
|
|
Eisenmenger Reaktion
|
Aus einem links-rechts-Shunt z.B. bei VDS wird im Laufe der Zeit ein rechts-links-Shunt bei fixierter PAH
|
|
|
Klinik von interstitiellen Lungenerkrankungen
|
1. Anhaltender trockener Husten
2. Belastungsdyspnoe Lufu: -Zuerst Einschränkung der Diffusionskapazität, im Verlauf restriktive Ventilationsstörung |
|
|
Irisblendenphänomen
|
Ein Fingerabdruck bleibt bei schwerer pAVK verlängert als blasse Stelle sichtbar.
|
|
|
Basisschema Kortison bei interstitiellen Lungenerkrankungen
|
Beginn mit 50mg/d Prednisolon
Nach 14 Tagen 40mg/d Danach monatlich um 10mg/d reduzieren bis zu einer Erhaltungsdosis von 5-15mg/d |
|
|
Leriche-Syndrom
|
Aortenbifurkationsverschluss (ischialgiforme Beschwerden, Erektionsschwäche)
|
|
|
Therapiebeurteilung bei interstitieller Lungenerkrankung
|
TLC
VC SaO2 Diffusionskapazität |
|
|
1. Pulsus celer et altus
2. Pulsus parvus et tardus |
1. Aortenklappeninsuffizienz ("Wasserhammerpuls")
2. Aortenklappenstenose |
|
|
Idiopathische pulmonale Fibrose (IPF)
|
-radiologisches Muster der usual interstitial pneumonia (UIP): Retikuläre Zeichnung mit honey combing in den basalen und mittleren Lungenabschnitten, subpleurale Betonung, Milchglastrübungen untypisch!
-schlechte Prognose, mittlere Überlebenszeit 3-6 Jahre -keine gesicherte Therapie -PG: Schaden an Typ II Pneumozyten -TBB führt oft zu einer akuten Exazerbation -Männer > 60J, Raucher -Listung zur Lungentransplantation! |
|
|
Morbus Binswanger
|
subkortikale arteriosklerotische Enzephalopathie
|
|
|
Nichtspezifische interstitielle Pneumonie (NSIP)
|
-häufig bei systemischen Grunderkrankungen (systemische Sklerose, rheumatoide Arthritis, medikamentös verursachte Lungenschädigungen)
-mitlere Überlebenszeit 10 Jahre -Durchschnittliches Erkrankungsalter 40-50 Jahre -CT: Basale retikuläre Zeichnungsvermehrung mit Milchglastrübungen, subpleurale Aussparungen, Traktionsbronchiektasen, deutlicher Volumenverlust des Unterlappens -Immer Therapieversuch mit Prednisolon, häufig gutes Ansprechen bis hin zur Komplettremission |
|
|
Erdheim-Gsell-Krankheit
|
Zystische Medianekrose, mit erhöhter Inzidenz für Aortendissektion assoziiert, Vorkommen bei angeborenen Bindegewebserkrankungen, insbes. Marfan Syndrom
|
|
|
Desquamative interstitielle Pneumonie (DIP)
|
-selten
-betrifft nur Raucher, überwiegend junge Männer -häufig Spontanremission nach Aufgabe des Inhalationsrauchens -Akkumulation von Rauchermakrophagen im Alveolarraum -CT: Milchglastrübungen -BAL: makroskopisch schmutzig braun wie die Finger eines starken Rauchers |
|
|
Erdheim-Gsell-Krankheit
|
Zystische Medianekrose, mit erhöhter Inzidenz für Aortendissektion assoziiert, Vorkommen bei angeborenen Bindegewebserkrankungen, insbes. Marfan Syndrom
|
|
|
Akute interstitielle Pneumonie (AIP)
|
=Hamman-Rich-Syndrom
Die AIP ist dem ARDS in jeder Hinsicht sehr ähnlich ("idiopathisches ARDS"). Schlagartiger Beginn, rasch progredienter Verlauf -Behandlungsversuch mit hochdosierten Steroiden (2 Tage 2x 500mg Prednisolon, 3 Tage 1x 500mg, dann 1 x 250mg), begleitend Antibiose und Antimykose |
|
|
Morbus Mondor
|
Idiopathische Thrombophlebitis der seitlichen Thoraxvenen, die als druckdolenter Strang tastbar sind, selbstlimitierender Verlauf
|
|
|
Lymphozytäre interstitielle Pneumonie (LIP)
|
-Reaktive Veränderung, meistens bei Systemerkrankungen
-Rö Thorax: Diffuse interstitielle Verdickungen v.a. basal aber auch herdförmige Verdichtungen mit positivem Bronchopneumogramm, CT: Zysten und Knötchen, kann Tumor imitieren. |
|
|
May-Thurner-Syndrom
|
Abflussbehinderung an der Kreuzungsstelle der linken Beckenvene und der rechten Beckenarterie mit Ausbildung einer septenartigen Leiste im Venenlumen = Venensporn (20% der Erwachsenen)
Kompl.: TVT des linken Beines |
|
|
Kryptogene organisierende Pneumonie (COP)
|
-früher BOOP
-Nichtraucher häufiger betroffen als Raucher -Infiltrate primär bds. peripher, typisches Wandern der Infiltrate -Allgemeinsymtome wie Schwäche, Gewichtsverlust, Nachtschweiß, Muskel- und Gelenkbeschwerden -spricht gut auf Kortikoide an. |
|
|
Virchow Trias
|
PG der TVT
1. Endothelalteration (Entz., Trauma) 2. Blutstromveränderung (Wirbel bei Varizen, lokale Stase) 3. Veränderung der Blutzusammensetzung (Thrombophilie) |
|
|
Sarkoidose
|
M. Boeck
-Epidem.: Schwarze Bevölkerung der USA, Schweden, Island -Radiologische Einteilung: Typ 0 - IV -Histo: Nicht verkäsende epitheloidzellige Granulome -Löfgren-Syndrom: Hochakute Form mit Erythma nodosum, Polyarthritis (Sprunggelenk) und bihilärer Lympadenopathie (junge Frauen) -Bei chronischer Verlaufsform Husten, Belastungsdyspnoe, Fatigue-Syndrom -Organbefall: Speicheldrüsen, Uveitis, Fascialisparese (Heerfordt-Syndrom), Knochen (M. Jüngling), Haut (Lupus pernio), Leber, Herz, Nervensystem -BAL: Lymphozytäre Alveolitis mit erhöhtem CD4/CD8 Quotienten (>5) Labor: IgG erhöht, BSG erhöht, Hyperkalzämie (erhöhte Produktion von 1,25-(OH2), evtl. Eosinophilie, Tuberkulin-Hauttest negativ, ACE und S-IL-2R = Aktivitätsparameter -Nach Therapie ist die Rezidivrate erhöht! |
|
|
Pel-Ebstein-Fieber
|
wellenförmiger Fieberverlauf bei M. Hodgkin
|
|
|
Radiologische Einteilung der Sarkoidose
|
Typ 0: Normalbefund
Typ I: Bihiläre Lymphadenopathie Typ II: Bihiläre Lymphadenopathie und Parenchymbefall Typ III: Parenchymbefall ohne Lymphadenopathie Typ IV: Lungenfibrose |
|
|
Mikulicz-Syndrom
|
Parotisschwellung und Tränendrüsenbefall bei CLL
|
|
|
Exogen allergische Alveolitis
|
Häufigste Auslöser sind Antigene biologischen Ursprungs, tpyisch sind die Farmerlunge und die Vogelhalterlunge, daneben aber auch viele andere. Auch: Chemische Stoffe wie Isozyanate (Chemiearbeiterlunge).
Kombinierte Typ III und Typ IV (zellgebundene) Hypersensitivitätsreaktion -Ausgeprägte lymphozytäre Alveolitis, starke Erhöhung der Lymphozytenzahl in der BAL (CD4/CD8 <1; normal 2,0) -Grippeähnliche Symptome treten 4-8 Stunden nach der Exposition auf. Abklingen der Beschwerden nach 24 Stunden. Danaben auch subakute chronische Form möglich. -Röntgenbild oft unauffällig, aber deutliche Diffusionsstörung (umgekehrt wie bei der Sarkoidose) -Akute Form reversibel, bei fortgesetzter Allergenexposition chronischer irreversibler Verlauf |
|
|
Gumprecht´sche Kernschatten
|
gequetschte Kerne von Lymphozyten bei CLL
|
|
|
Progressive systemische Sklerose
|
Kollagenanhäufung und Fibrose von Haut und inneren Organen + obliterierende Angiopathie
Hautveränderungen (100%): 3 Stadien: Ödem -> Induration -> Atrophie Sekundäres Raynaud-Syndrom Sklerodaktylie, Rattenbissnekrosen, mimische Starre des Gesichtes, Mikrostomie, Tabaksbeutelmund Arthritis/ Arthralgien (50-70%) Organmanifestationen: -Sleroglossom, Motilitässtörung des Ösophagus (Weitstellung der distalen 2/3), Wassermelonenmagen (80%) -Häufig Lungenfibrose (Bild der NSIP), pulmonale Hypertonie (v.a. bei CREST-Syndrom) (20-70%) -Myokarditis, Myokardfibrose (20%) -PAH (15%, v.a. bei CREST-Syndrom) -Niereninfarkte, Mikroangiopathie (20%) Akrale limitierte Verlaufsform: CREST-Syndrom (Calcinosis cutis, Raynaud, Ösophagusbeteiligung, Sklerodaktylie, Teleangiektasien), häufig PAH! Im CT: Basale interstitielle Verdichtungen und dilatierter Ösophagus ANA (90%), ACA (70% bei CREST) |
|
|
Stadien art. Hypertonie
|
Optimal <120/80mmHg
Normal 120-129/80-84mmHg hoch-normal 130-139/85-89mmHg HPT Grad1 140-159/90-99mmHg HPT Grad 2 160-179/100-109mmHg HPT Grad 3 >=180/110 mmHg |
|
|
Polymyositis/ Dermatomyositis
|
Entzündliche Systemerkrankung der Skelettmuskulatur mit lymphozytärer Infiltration
->Myositis der proximalen Extremitätenmuskulatur (Schwierigkeiten beim Aufstehen und Heben der Arme über die Horizontale, Myalgien) ->Hautveränderungen: Lividrote, ödematöse Erytheme des Gesichtes, bes. periorbital (lila Ringe), lichenoide weißliche bis blassrote Papeln der Fingerstreckseiten (Gottron-Papeln), druckschmerzhafte Nagelfalzhyperkeratosen (Keinig-Zeichen), Erytheme und Rhagaden der Handflächen (Mechanikerhände) -Organbeteiligung: Ösophagus (Schluckstörungen), Herz (Myokarditis), Lunge (Fibrose) CK, GOT LDH Erhöhung, BSG Erhöhung, ANA Elektromyogramm, MRT (Ödem), Muskelbiopsie Tumorsuche! Th.: Steroide, Immunsuppressiva |
|
|
Differentialdiagnose Muskelschwäche/ Schmerzen
|
-Polymyositis, Dermatomyositis
-Medikamenten-induziert (CSE-Hemmer) -Alkoholmyopathie -Polymyalgia rheumatica -Muskeldystrophie -Myasthenia gravis -Lambert Eaton Syndrom -Infektiöse Myositis (Caxsackie, Trichinen) |
|
|
Auerstäbchen
|
Struktur im Zytoplasma bei Myeloblasen-Leukämie
|
|
|
Kollagenosen
|
1. Systemischer Lupus erythematodes
2. Polymyositis, Dermatomyositis 3. Sklerodermie 4. Sjögren-Syndrom 5. Sharp-Syndrom (Mischkollagenosen) |
|
|
Pseudo-Gaucher-Zellen
|
prognostisch günstiges Zeichen bei CML
|
|
|
Vaskulitiden
|
Kleine Gefäße:
-M. Wegener (ANCA) -Churg-Strauss-Syndrom (ANCA) -Mikroskopische Panarteriits (ANCA) -Purpura Schoenlein Hennoch -Vaskultis bei essentieller Kryoglobulinämie Mittlere Gefäße -Klassische Panarteriitis -M. Kawasaki Große Gefäße -Riesenzell-Arteriitis -Takayasu-Arteriitis |
|
|
Gumprecht´sche Kernschatten
|
gequetschte Lymphozytenkerne bei CLL
|
|
|
Cholera
|
Vibrio Cholerae/ El Tor: gekrümmte gramnegative Stäbchen mit monotrich polarer Begeißelung
Choleratoxin Massenerkrankung bei Armut, Mangelernährung und niedrigem Hygiene-Standard Magensäure wirksamer Schutz Zielzellen: Epithel des Dünndarms -> sekretorische Diarrhoe (Toxin bewirkt Erhöhung des cAMP-Spiegels ->Hypersekretion, Verlust von bis zu 25l Flüssigkeit/d) Klinik: nur 15% der Infektionen symptomatisch, Verlauf sehr variabel, Reiswasserstuhle mit Exsikkose Di.: Wattetupfer Rektal/Stuhlabstrich, 1% Peptonlösung (Vibrionen gehen bei Austrocknung rasch zu Grunde) Therapie: Substitution von Wasser und Elektrolyten; Makrolid oder Chinolon Lebendimpfstoff und Totimpfstoff vorhanden Inf. mit Vibrio El Tor verläuft weniger dramatisch als mit Vibrio cholerae |
|
|
Hedinger Syndrom
|
Kardiale Manifestation des Karzinoid-Syndroms: Endokardfibrose, bevorzugt des rechten Herzens und evtl. dadurch bedingte TI, evtl. PS
|
|
|
WHO Salz- Glukoselösung
|
20g/l Glucose
2,5g/l Natriumbicarbonat 3,5g/l Natriumchlorid 1,5g/l Kaliumchlorid |
|
|
Schellong Test
|
Stehversuch:
10 Min. Liegen 10 Min. Stehen Normal: RR Abfall systol. <20mmHg, diastol. < 10mmHg |
|
|
Qurantäneerkankungen der WHO
|
Cholera
Pest Gelbfieber |
|
|
Fitz Hugh Curtis Syndrom
|
Perihepatitis nach Gonokokken- oder Chlamydieninfektion
|
|
|
Meldepflichtige Krankheiten
|
Adenovirus Konjunktivitis, Botulismus, Brucellose, Cholera, Diphtherie, Echinokokkose, Enteritis durch einige Erreger, HUS, Fleckfieber, FSME, Haemophilus influenzae, HIV (nichtnamentlich), Creutzfeld-Jakob, Vogelgrippe, Legionellose, Lepra, Leptospirose, Listeriose, Lues, Malaria, Masern, Meningokokken, Milzbrand, Ornithose, Paratyphus, Pest, Polio, Psittakose, Q-Fieber, konnatale Röteln, Shigellen, Tollwut, konnatale Toxoplasmose, Trichinose, Tuberkulose, Tularämie, Typhus, virusbed. hämorrh. Fieber (Ebola, Gelbfieber, Hanta, Lassa, Marburg), Virushepatitis
|
|
|
Caplan Syndrom
|
Kombination von Silikose und chronischer Polyarthritis
|
|
|
Meldepflichtige Durchfallerkrankungen
|
Adenoviren, Astroviren, Coronaviren, Noroviren, Rotaviren
Campylobacter, E. coli, Salmonellen, Shigellen, Yersinien Entamoeba histolytica, Giardia lamblia |
|
|
Osteodystrophiea cystica generalisata von Recklinghausen
|
"Braune Tumoren": eingeblutete Resorptionszysten in den Knochen bei PHPT
|
|
|
Systemischer Lupus erythematodes
|
Vaskulitis/ Perivaskulitis der kleinen Arterien und Arteriolen sowie Ablagerung von Immunkomplexen
SLE-Kriterien (bei 4 Kriterien Lupus wahrscheinlich): 1. Schmetterlingserythem 2. Diskoider Lupus erythematodes 3. Fotosensibilität 4. Orale und nasale Schleimhautulcera 5. Nichterosive Arthritis von 2 oder mehr Gelenken 6. Serositis (Pleuritis, Perikarditis) 7. Nierenbeteiligung 8. ZNS-Beteiligung 9. hämolytische Anämie, Thrombopenie, Leukopenie 10. Anti-ds- DNS, Anti-Sm, Antiphospholipidantikörper 11. ANA Man unterscheidet eine kutane Form und eine systemische Form (kutane Form: günstige Prognose) Hautbiopsie: Lupusband (Ablagerungen entlang der Basalmembran) Wichtig bei allen Formen: Lichtschutz! medikamentös induzierter Lupus: Procainamid, Hydralazin, Methyldopa, Phenytoin, Neuroleptika, Etanercept, Infliximab |
|
|
Mattglas-Effekt
|
Osteopenie des Schädels bei PHPT
|
|
|
Einteilung Vorhofflimmern (ACC/ AHA, ESC 2006)
|
1. Paroxysmal (selbstlimitierend, kürzer als 7 Tage, gewöhnlich <48h)
2a. Persitierend (terminiert durch Kardioversion, meist länger > 7 Tage) 2b. Langandauernd persistierend (>= 1 Jahr) 3. Permanent (akzeptiert anhaltend, kann nicht mit Kardioversion beendet werden oder Kardioversion wurde nicht versucht.) Mikro-Reentry im Mündungsbereich der Pulmonalvenen, Vorhoffrequenz 350-600/min, Filterfunktion des AV-Knotens Meist schmale QRS-Komplexe, durch Ashman-Phänomen teils verbreiterte QRS-Komplexe (aberrierende Leitung) |
|
|
Horner Syndrom
|
Miosis
Ptosis Enophthalmus (Läsion des Halssympthikus) |
|
|
Ursachen für Vorhofflimmern
|
Hyperthyreose
hypertensive Herzkrankheit! chronischer Alkoholabusus Ausdauersport Schlafapnoe-Syndrom Typ-A-Verhalten "lone atrial fibrillation" genetische Präsdisposition assoziiert sind: art. Hypertonie (!), Diabetes mellitus, Herzklappendefekte, Herzinsuffizienz |
|
|
Sipple Snydrom
|
MEN 2a
-C-Zellkarzinom -Phäochromozytom -primärer Hyperparathyroidismus |
|
|
CHADS2-Score
|
C Cardiac failure
H Hypertension A Age (>75J) D Diabetes mellitus S Stroke INR 2-3 Neu: CHA2DS2-Vasc-Score V Vascular Disease A Age 65-74 Jahre Sc Weibliches Geschlecht Maximal 9 Punkte Fazit: Die OAK wird fast immer epmfohlen, Ausnahme: Männer <65J. |
|
|
Gorlin Sydrom
|
MEN 2b
C-Zellkarzinom -Phäochromozytom -Schleimhautneurinome und marfanoider Habitus |
|
|
primärer Hyperparathyroidismus
|
85% Adenome, 15% Hyperplasie der Epithelkörperchen
"Stein-, Bein- und Magenpein" -Neprholithiasis, Niereninsuffizienz -Osteopenie "Mattglaseffekt des Schädels" -Übelkeit, Obstipation, Ulcera ventriculi, Pankreatitis -Muskelschwäche -Depression Hyperkalzämische Krise (hohe Letalität): -Polyurie, Polydipsie -Erbrechen, Exsikkose -Koma -Herzrhyhtmusstörungen Durch die rasche Entwicklung einer Niereninsuffizienz mit Anstieg des Serumphosphats können Kalzifizierungen in verschiedenen Organen auftreten (z.B. Kornea, Media der Arterien), Herzrhyhmusstörungen können zum plötzlichen Tod führen. Therapie: ggf. operativ |
|
|
Riedel Struma
|
seltene Form der chronischen Thyreoiditis, chronisch fibrosierend
|
|
|
Differentiialdiagnosen bei Hyperkalzämie
|
-pHPT: PTH hoch
-Tumor: PHT niedrig, bei manchen TU PTHrP hoch (Parathormon-verwandtes Peptid) -Sarkoidose: Vit D hoch |
|
|
Complete linkage
|
the inheritance patterns for 2 genes on the same chromosome when the observed frequency for crossover between the loci is 0.
|
|
|
Wirkungen von
PHT Vit D Calcitonin |
PTH: Erhöhung des Calciumspiegels im Blut (Aktivierung von Osteoklasten, Erhöhung der Calcium-Rückresorption in der Niere)
Vit D: Steigerung der enteralen Calcium und Phosphat Aufnahme, Mineralisierung des Knochens Calcitonin: Gegenspieler des PTH -> Mineralisierung des Knochens, Steigerung der Rückresorption in der Nieren |
|
|
Darymple Zeichen
|
sichtbarer Sklerastreifen oberhalb der Hornhaut beim Blick geradeaus bei endokriner Orbitopathie
|
|
|
Pathogenese des Hyperkalzämie bei Tumoren
|
1. Knochenmetastasen (Stimulierung der Osteoklasten über Zytokine)
2. Paraneoplastische Hyperkalzämie (durch ektope Bildung parathormonverwandter Peptide PTHrP 3. Ektope Hydroxylierung von Vit D zu Calcitriol (Lymphome) |
|
|
Graefe Zeichen
|
Zurückbleiben des Oberlides bei Blicksenkung bei endokriner Orbitopathie
|
|
|
Therapie der hyperkalzämischen Krise
|
-forcierte Diurese
-Bisphosphonate -Bei Vit D vermittelter Hyperkalzämie Glukokortikoide (Sarkoidose, Lymphome) |
|
|
Möbius Zeichen
|
Konvergenzschwäche bei endokriner Orbitopathie
|
|
|
Klinik der Hyperkalzämie
|
-Polyurie, Polydipsie, Niereninsuffizienz
-Übelkeit, Erbrechen, Obstipation -Muskelschwäche -Herzrhyhtmusstörungen -Somnolenz, Koma, Psychose |
|
|
Marine-Lenhart-Syndrom
|
Kombination von Schiddrüsenautonomie und M. Basedow
|
|
|
Pathogenese der Niereninsuffizienz bei Hyperkalzämie
|
-Volumendepletion (distaler Tubulus verliert Konzentrationsfähigkeit)
-renale Vasokonstriktion |
|
|
Whipple Trias
|
Hypoglykämie
1. BZ<45mgdl 2. hypoglykämische Symptome 3. Verschwinden der Symptome unter Glukosegabe |
|
|
Los Angeles Klassifikation der Refluxösophagitis
|
A Eine oder mehrere Erosionen < 5mm Durchmesser, die sich nicht zwischen den Kuppen der Mukosaflaten erstrecken
B wie A aber Ersionen >= 5mm C Erosionen erstrecken sich zwischen 2 oder mehr Kuppen der Mukosafalten, erfassen aber < 75% der Zirkumferenz D Wie C aber >= 75% der Zirkumferenz |
|
|
Mennel-Zeichen
|
Kreuschmerzen, wenn beim seitlich liegenden Patienten das untere Bein maximal gebeugt, das andere retrofelktiert wird
M. Bechterew |
|
|
5 Hauptformen der Kardiomyopathien
|
1. DCM
2. HCM 3. RCM 4. ARVD 5. Nicht klassifizierbare (Tako Tsubo, NCCM) |
|
|
Schober-Maß
|
Die im Stehen gemessene Distanz vom 5. LWK 10cm nach kranial muss sich bei maximaler Rumpfbeugung um mindestens 4 cm vergrößern.
|
|
|
Nephrotisches Syndrom
|
1. Proteinurie > 3 - 3,5 g/d
2. Hypoproteinämie (evtl. Infektanfälligkeit durch IgG-Mangel) 3. Hypalbuminömische Ödeme 4. Hyperlipoproteinämie mit Erhöhung von Cholesterin und Triglyceriden Häufigste Ursachen: Glomerulonephritis, diabetische Nephropathie, Plasmozytom Serumelektrophorese: Vermindert Albumin und Gamma-Globuline; relative Zunahme von Alpha- und Beta-Globulinen Infektanfälligkeit durch IgG-Verlust! Thrombembolien durch AT-Verlust! |
|
|
Off-Maß
|
Die im Stehen gemessene Distanz vom 7. HWK 30cm nach kaudal muss sich nach maximaler Rumpfbeugung um mind. 2cm vergrößern.
|
|
|
Churg-Strauss-Syndrom
|
Granulomatöse, nicht nekrotisierende Vaskulitis mit eosinophilen Infiltraten
ACR-Kriterien: 1. Asthma bronchiale 2. Flüchtige pulmonale Infiltrate 3. Sinusitiden 4. Eosinophilie >10% 5. Polyneuropathie 6. Biopsie: Nachweis einer extravaskulären Eosinophilie Typisch ist auch eine kardiale Beteiligung mit Koronaritis oder Myokarditis; ZNS-Vaskulitis Labor: Eosinophilie, eröhtes IgE 5-Jahres-ÜR: 60% |
|
|
Bambusrohrform der Wirbelsäule
|
Verkalkungen des Wirbelkörperbandapparates bei M. Bechterew im Endstadium
|
|
|
Purpura Schoenlein-Henoch
|
Immunreaktion vom Typ III mit Ablagerung von IgA-haltigen Immunkomplexen in kleinen Gefäßen und Aktivierung des Komplementsystems. Zeitlicher Zusammenhang mit einem Infekt der oberen Luftwege (50% Influenza A)
Meist Kinder im Vorschulalter 1. Petechien ("tastbare Purpura") 2. Sprunggelenksarthritis 3. Kolikartige Bauchschmerzen (vaskulitisch bedingte Darmischämien) 4. Mesangioproliferative Glomerulonephritis 5. Kopfschmerzen, Verhaltensstörungen Di.: -Nachweis zirkulierender Immunkomplexe -IgA erhöht -Hautbiopsie |
|
|
Balanitis circinata
|
Psoriasiforme Erytheme der männlichen Genitalschleimhaut im Rahmen eines Reiter-Syndroms
|
|
|
Morbus Behcet
|
Leukoklastische Vaskulitis mit Befall von Arterien und Venen
Orale Aphten, genitale Aphten Augenbeteiligung Arthritis GIT, ZNS |
|
|
Keratoderma blenorrhagicum
|
Pustulöse Veränderungen an Handflächen und Fußsohlen im Rahmen eines Reiter-Syndroms
|
|
|
Akute eosinophile Pneumonie
|
gehäuft bei Jugendlichen, die ihre ersten Zigaretten rauchen -> vermutlich überschießende Immunreaktion auf Substanzen im Zigarettenrauch
Klinisch wie schwere akute Lobärpneumonie, schwere Hypoxmie typisch, ARDS Therapie: Glukokortikoide! gut behandelbar |
|
|
Herbeden´ Knoten
|
Erosive Polyarthrose der Fingerendgelenke
|
|
|
Chronisch eosinophile Pneumonie
|
gehäuft bei rauchenden Frauen
Subakuter Verlauf mit subfebrilen Temperaturen, Gewichtsverlust, Nachtschweiß, asthmatischen Symptomen Radiologisch: Periphere bilaterale Infiltrate, Negativbild eines Lungenödems Im Blutbild ausgeprägte Eosinophilie Therapie: Glukokortikoide über längeren Zeitraum |
|
|
Bouchard´ Knoten
|
erosive Polyarthrose der Fingermittelgelenke
|
|
|
Eosinophile Lungenparenchymerkrankungen
|
-Akute eosinophile Pneumonie
-Chronische eosinophile Pneumonie -Churg-Strauss-Syndrom -Löffler-Infiltrat bei Lugenpassage von Nematoden -medikamenteninduziert (ACE-Hemmer, Amiodaron) -allergische bronchopulmonale Aspergillose -Toxiniduziert (Toxic-Oil-Syndrom) |
|
|
Gaensslen-Zeichen
|
Querdruckschmerz der Hand bei RA
|
|
|
Strahlenpneumonitis
|
Werden über ein Drittel des Lungenvolumens mit Dosen > 20Gy bestrahlt, entwickelt sich in einem Drittel eine Strahlenpneumonitis. Eine gleichzeitige Chemotherapie erhöht das Risiko.
Therapie: Glukokortikoide, ggf. auch zur Prophylaxe |
|
|
Phalen-Zeichen
|
Auslösung von Schmerzen bei Karpaltunnelsyndrom durch maximale Beugung oder Streckung im Handgelenk
|
|
|
Allergische bronchopulmonale Aspergillose
|
Vorkommen bei Asthmapatienten und Mukoviszidose-Patienten
ABPA wahrscheinlich bei folgenden Kriterien: -Asthmaerkrankung -Sofortreaktion auf Aspergillus im Hauttest -erhöhtes spezifisches IgE und IgG gegen Aspergillus-Species -erhöhtes Gesamt IgE -zentrale Bronchiektasen Ätiologie: Inhalativ erworbene Besiedlung mit Aspergillussporen; im Mukus, nicht im Gewebe Zäher Mukus, der Bronchien okkludiert Therapie: Steroide, bronchoskopische Entfernung der Mukusplaques |
|
|
Hoffmann-Tinel-Test
|
Auslösung von Schmerzen durch Beklopfen des Karpaltunnels bei Karpaltunnelsyndrom
|
|
|
Lungeninfiltrate durch Medikamente
|
-Bleomycin
-Methotrexat (Diabetes erhöhet Toxizität!) -Amiodaron Risiko für Lungenfibrose korreliert mit gleichzeitiger Gabe von Sauerstoff |
|
|
Caplan-Syndrom
|
RA + Silikose
|
|
|
TRALI
|
Transfusion-related acute lung injury
Innerhalb von 1-2 Stunden nach Gabe von FFP (selten EK) auftretendes Krankheitsbild mit Dyspnoe, Hypoxämie, Lungeninfiltraten. Gehäuft bei Sepsis. DD Lungenödem |
|
|
Felty-Syndrom
|
Schwere Verlaufsform der RA im Erwachsenenalter
|
|
|
Rapid progressive Glomerulonephritis
|
RPGN
Typ I (10%): Antibasalmembran-RPGN (mit oder ohne Lungenbeteiligung - Goodpasture Syndrom) Typ II (40%): Immunkomplex-RPGN (postinfektiös, SLE, Schoenlein-Hennoch) Typ III (50%): ANCA-assoziierte Vaskulitiden (mPA, M. Wegener) Nephrotisches Syndrom Immunsuppressive Behandlung, evtl. Plasmaaustausch |
|
|
Morbus Still
|
Systemische Form der RA bei Kindern, selten auch bei Erwachsenen
|
|
|
Def. Hämatothorax
|
Ist der Hämatokrit im Erguss über der Hälfte des Bluthämatokrits, spricht man von einem Hämatothorax.
|
|
|
Katayama-Syndrom
|
Fieber, Husten, Eosinophilie bei Bilharziose (Lungenpassage)
|
|
|
Exsudat - Transsudat
Pleuraempyem |
Light-Kriterien für Exsudat:
1. Proteingehalt > 3g/dl 2. LDH > 0,5-0,6 des oberen LDH-Sollwertes (>200U/l) (3. Cholesterin > 45mg/dl) Bei Eindickung des Blutes (z.B. Diuretikatherapie) oder Überwässerung kann die Grenze verwischt werden. Pleuraempyem: pH < 7,0, Leukos vermehrt (Azidose durch Laktat, das beim Abbau von Glucose durch Bakterien entsteht) |
|
|
Goldblatt-Effekt
|
Aktivierung des RAAS bei Nierenarterienstenose mit konsekutiver art. Hypertonie
|
|
|
Differentialdiagnose Pleuraerguss Transsudat
|
1.Herzinsuffizienz
2. Hypoproteinämie -Nephrotisches Syndrom -Leberzirrhose -Enterales Eiweißverlustsyndrom |
|
|
Renovaskuläre Hypertonie
|
1% aller Hypertonien
einseitige oder beidseitige Nierenarterienstenose 75% arteriosklerotisch 25% fibromuskulär (jüngere Patienten) Goldblatteffekt (Aktivierung des RAAS) Klinik: -schwer einzustellende art. Hypertonie -Strömungsgeräusch -flash pulmonary edema -Verschlechterung der Nierenfunktion unter ACE-Hemmer oder AT1-Blocker Di.: -Farbdoppler: Vmax A. renalis >=2m/s -CT, MRT, DSA |
|
|
Rheumatisches Fieber - Jones Kriterien
|
Hauptkriterien:
1. Karditis 2. Wandernde Polyarthritis 3. Chorea minor 4. Subkutane Knötchen 5. Erythema anulare rheumaticum Nebenkriterien: 1. Fieber 2. Arthralgie 3. BSG u./o. CRP Erhöhung 4. Verlängerte PQ- oder PR-Zeit |
|
|
Bei welchen Tumoren muss man an die Möglichkeit eines MEN denken?
|
1. medulläres Schilddrüsenkarzinom
2. endokriner Pankreastumor 3. Phäochromozytom 4. pHPT |
|
|
Rheumatisches Fieber Pathomechanismus
|
Beginn 2 Wochen nach einer Tonsillopharyngitis mit Beta-hämolysierenden Streptokokken
Das typenspezifische M-Protein der S. zeigt eine Kreuzreaktivität mit den sarkolemmalen Antigenen Tropomyosin und Myosin |
|
|
Cullen-Zeichen
|
bläuliche Flecken periumbilikal bei hämorrhagischer Pankreatitis
|
|
|
Therapie rheumatisches Fieber
|
Penicillin V
ASS Kortikosteroide Rezidivprophylaxe mit Penicillin über mindestens 10 Jahre, max. bis 25.LJ |
|
|
Grey-Turner-Zeichen
|
bläuliche Flecken im Flankenbereich bei hämorrhagischer Pankreatitis
|
|
|
OP-Indikation bei Mitralinsuffizienz
|
Symptomatische Patienten:
EF > 30% (<30% nur Rekonstruktion) Asymptomatische Patienten: EF < 60% und/ oder LVEDD > 45mm Vorhofflimmern pPA > 50mmHg EF > 60% und LVEDD >45mm aber fehlende kontraktile Reserve |
|
|
Jaccoud-Arthropathie
|
Polyarthritis ohne Erosionen, evtl. aber mit Subluxationen/ Fehlstellungen bei SLE
|
|
|
Glutensensitive Enteropathie
|
Unverträglichkeit gegenüber der Gliadinfraktion des Gluten (Weizen, Gerste, Roggen, Dinkel, Grünkern)
Kl.: 1. Klassische Zöliakie mit Durchfällen, Gewichtsverlust und Malabsorptionssyndrom 2. Atypische Verläufe: Dermatitis herpetiformis Duhring, Eisenmangelanämie, Osteoporose, chron. Hepatitis, aymptomatische Sprue 3. Erhöhtes Risiko für T-Zelllymphome des Dünndarms Labor: AGA (Anti-Gliadin-AK) AEA (Anti-Endomysium-AK) ATG (Anti-Transglutaminase-AK) (am spezifischsten) ESPGAN-Kriterien 1. Histologie (Marsh-Kriterien mit Zottenatrophie und Kryptenhyperplasie, Vermehrung intraepithelialer Lymphozyten) 2. Klinische Besserung unter glutenfreier Diät Oft sekundärer Laktasemangel Th.: Glutenfreie Diät mit Kartoffeln, Mais, Reis, Hirse, Soja |
|
|
Nephritisches Syndrom
|
Das nephritische Syndrom ist ein im Rahmen der Schädigung von Glomeruli auftretendes klinisches Syndrom.
Volhard-Trias: * Hämaturie * Ödeme (betont an Augenlidern) infolge einer Wasserretention * Hypertonus Daneben tritt häufig Flankenschmerz auf. Bei komplizierten Verläufen kommt es zur Ausbildung eines Lungenödems (Dyspnoe) und einer Oligurie bzw. Anurie. |
|
|
Was dürfen Patienten mit Sprue essen?
|
Kartoffeln
Mais Reis Hirse Soja |
|
|
Nephritisches Syndrom - Nephrotisches Syndrom
|
Nephritisches Syndrom:
Volhard-Trias: 1. Hämaturie 2. Ödeme (Augenlider) 3. Hypertonus Nephrotisches Syndrom 1. Starke Proteinurie 2. Hypoproteinämie 3. Hypalbuminämie 4. Hyperlipoproteinämie |
|
|
Kolonpolypen
|
-entzündlicher Polyp
-hyperplastischer Polyp -Harmatom -Adenom Adenome: -Klassisches Adenom, breitbasig oder gestielt, histologisch tubulär, tubulo-villös, villös -Sessiles serratiertes Adenom (SSA), kaum über Schleimhautniveau -Traditionell serratiertes Adenom, makroskopisch wie klassisches Adenom -gemischtes Adenom |
|
|
Heyde-Syndrom
|
Assoziation von Aortenklappenstenose und von Willebrand-Jürgens-Syndrom mit GI-Blutungen
|
|
|
Wann wird eine Kolonneoplasie als invasives Karzinom klassifiziert?
|
Bei Überschreiten der Muscularis Mucosae
|
|
|
Ghon-Herd
|
Primärkomplex der Tbc= Ghon-Herd + lokale Lymphknotenreaktion
|
|
|
Hereditäre Polyposis Syndrome
|
1. FAP (Familiäre adenomatöse Poliposis), autosomal dominant erblich, Mutation des APC-Tumorsuppressorgens, obligate Präkanzerose
2. Conkhite-Canada-Syndrom: Generalisierte Polypose des GIT, bräunliche Hautpigmentierung, Alopezie, Diarrhoe, kein erhöhtes Karzinomrisiko 3. Peutz-Jeghers-Syndrom: Polyposis des Dünndarms (Harmatome), Melaninflecken an Lippen und Mundschleimhaut, Stieldrehung der Polypen, erhöhtes Risiko für Pankreas Karzinome und Ovarial-Karzinom |
|
|
Simon-Spitzenherd
|
Tbc: Im Rahmen der Primärinfektion können auf dem Blutwege kleine Organherde entstehen. In den Spitzenfeldern der Lunge: Simon´ Spitzenherde)
|
|
|
Nachsorge bei sporadischen kolorektalen Polypen
|
-unauffällig oder 1 hyperplastischer Polyp -> 10 Jahre
- 1 bis 2 klassische tubuläre Adenome < 10mm ohne HG-IEN -> 5 Jahre - 3 bis 10 klassische tubuläre Adenome ohne HG-IEN oder 1 > 10mm oder 1 villöses A. oder 1 Adenom mit HG-IEN oder 1 serratiertes Adenom -> 3 Jahre -mehr als 10 Adenome -> < 3 Jahre |
|
|
Landouzy-Sepsis
|
Tbc-Sepsis
|
|
|
Extrakolische Manifestationen der FAP
|
-Adenome des Duodenums und Drüsenkörperzysten im Magen (Risiko für Duodenalkarzinome)
-Epidermoidzysten und Osteome (Gardner-Syndrom) -Glio-/Medulloblastome (Turcot-Syndrom) -Congenitale Hypertrophie des retinalen Pigmentepithels (harmloser Augenhintergrundsbefund) |
|
|
Assmann-Frühinfiltrat
|
Reaktivierung eines alten Tbc-Spizenherdes, meist infra- und retroklavikulär gelegen.
|
|
|
Corrigan-Zeichen
|
Sichtbare Pulsation der Carotiden bei AI
|
|
|
Stadien Kolonkarzinom
|
TNM
TIS intraepithelial oder Infiltration der Lamina propria T1 Submukosa T2 Muscularis propria T3 Subserosa T4a viszerales Peritoneum T4b andere Organe/ Strukturen N je nach Anzahl der LK M1a Fernmetastasen in 1 Organ M1b Fernmetastasen in > 1 Organ oder Peritoneum Stadium 0: TIS Stadium I: T1, T2 Stadium IIA: T3 N0 M0 Stadium IIB: T4a N0 M0 Stadium IIC: T4b Stadium IIIA: T1,T2,N1a oder T1 N2a Stadium IIIB T3, T4a N1 oder T2, T3 N2a oder T1, T2 N2b Stadium IIIC: T4a, N2a oder T3, T4b N2b oder T4b, N1, N2 Stadium IVA: Jedes T, jedes N, M1a Stadium IVb: Jedes T, jedes N, M1b |
|
|
Quincke-Zeichen
|
Sichtbarer Kapillarpuls nach leichtem Druck auf einen Fingernagel bei AI
|
|
|
Operationsverfahren bei Rektum- /Kolonkarzinomen
|
Rektumkarzinom: anteriore Rektumresektion mit totaler Mesorektumresektion (TME)
Grenze der kontinenzerhaltenden Chirurgie liegt bei 5cm ab Anokutanlinie Kolonkarzinom: Hemikolektomie rechts oder links, Kolon-Transversumresektion, Sigmaresektion |
|
|
de Musset Zeichen
|
pulssynchrones Kopfnicken bei AI
|
|
|
Häufigkeiten der Lokalisation kolorektaler Karzinome
|
50% Rektum
30% Sigma 10% Coecum/ Colon ascendens 10% übriges Kolon |
|
|
Wasserhammerpuls
|
Pulsus celer et altus bei AI
|
|
|
Adjuvante Therapie beim Kolorektalen Karzinom
|
Stadium III
FOLFOX (Oxaliplatin + 5-FU + Folinsäure) -> Verbesserung der 5-Jahresüberlebensrate um 15-20% ebenso werden verwendet: XELOX (Capecitabin + Oxaliplatin) FOLFIRI (statt Oxaliplatin Irinotecan) ASS ! |
|
|
Morbus Simmonds
|
Totaler Ausfall des HVL mit klinischem Vollbild
|
|
|
Monoklonale Antikörper zur Behandlung von kolorektalen Karzinomen, Prognosemarker
|
Anti-EGFR (Epidermal Growth Factor Receptor): Cetuximab (Erbitux), Panitumumab (Vectibix); Prognosemarker: Hauttoxizität positiv, KRAS-Wildtyp positiv
Anti-VEGF (Vascular Endothelial Growth Factor): Bevacizumab (Avastin) |
|
|
Herxheimer Reaktion
|
Bei Behandlung einer Lues Reaktion durch Zerfallsprodukte:
Fieber, Myalgien, Kopfschmerzen, Hypotonie |
|
|
Prognoseabschätzung bei Lebermetastasen bei KRK
|
Fong-Score <= 2: gute Prognose
-nodal pos. Primärtumor -krankheitsfreies Intervall < 12 Monate -Metastasengröße < 5cm -mehr als 1 Metastase -CEA > 200ng/dl |
|
|
Kimmelstiel-Wilson
|
diabetische Glomerulosklerose
|
|
|
Häufigste angeborene Herzfehler im Erwachsenenalter
|
VSD (35%)
ASD (25%) Pulmonalstenose (10%) Aortenisthmusstenose (8%) |
|
|
VSD
|
Häufigster angeborener Herzfehler beim Erwachsenen (35%).
Kleine bis mittelgroße VSD wirken drucktrennend, bei großen Defekten kommt es zum Druckangleich. Volumenbelastung von LA und LV und Lungengefäßen. Bei großem VSD im Laufe von Jahren Eisenmenger-Reaktion. Verschlussrate bis zum 7. LJ hoch Klinik bei größeren VSD: Rechtsherzbleastung (PAH), rez. bronchopulmonale Infekte, Aorteninsuffizienz beim suprakristalen Defekt, Endokarditis, Arrhyhtmien und Blockbilder; nach Eisenmenger-Reaktion Zyanose mit Uhrglasnägeln und Trommelschlegelfingern Niedriger Blutdruck mit kleiner Amplitude, Systolikum (verschwindet beim großen Defekt) |
|
|
ASD
|
ASD II (80%, Ostium secundum Defekt, Bereich der Fossa ovalis)
ASD I (15%, Ostium primum Defekt) Bei Erwachsenen 25% der angeborenen Vitien Links-rechts-Shunt mit großem HZV im kleinen Kreislauf, sekundäre Widerstanderhöhung im Lungenkreislauf (im weiteren Verlauf Eisenmenger-Reaktion) Bei kleinem ASD bis 80% Spontanverschluss bis 4.LJ. Symptome oft erst nach dem 40. LJ, nach dem 60. LJ nahezu alle Patienten symptomatisch. Patienten mit kleinem Defekt werden spät symptomatisch. Kl.: Vorhofflimmern, paradoxe Embolien, Rechtsherzinsuffizienz, PAH, rezidivierende bronchopulmonale Infekte Verschlussindikation bei allen symptomatischen Kindern und jungen Erwachsenen, KI bei zu hohem pulm Widerstand |
|
|
Marfan-Syndrom
|
Mutation im Fibrillin-Gen auf Chromosom 15, autosomal dominant erblich, 25% Neumutationen
Klinik: -überlange Gliedmaßen, große Körperlänge, Arachnodaktylie, überdehnbare Gelenke, schmaler Kiefer mit schiefen Zähnen, Trichter- oder Kielbrust -Aortenaneurysmen (v.a. Aorta ascendens), AI, MI, TI -Kurzsichtigkeit, Netzhautablösung Besonders in der aszendierenden Aorta progrediente Mediadegeneration mit Risiko Aortenaneurysma und Ruptur! Prophylaxe: Betablocker, AT-Blocker. OP ab 45mm, bei pos. Fam-Anamnese für Ruptur ab 40mm. A. desc. > 55mm. Endokarditisprophylaxe! Mögl. systolische/ diastolische Herzinsuffizienz, ventr. Arrhythmien |
|
|
Schartz-Bartter-Syndrom
|
SIADH (Syndrom der inadäquaten ADH-Ausschüttung)
Ät.: paraneoplastisch (Kleinzeller), Legionellen-Pneumonie, medikamentöse, idiopathisch Klinik: -Appetitlosigkeit, Übelkeit, Erbrechen -Kopfschmerzen, Reizbarkeit, Persönlichkeitsveränderung, Stupor -Keine Ödeme, da die Wasserretention nur 3-4l beträgt -Hyponatriämie, hypertoner Urin (>300mOsm/kg) Therapie: Flüssigkeitsrestriktion, evtl. Vaptane, ggf. hypertones NaCl |
|
|
Vaptane
|
Vasopressin-Rezeptor-Antagonisten
z.B. Tolvaptan Aquarese mit Anstieg des Serumnatriums |
|
|
pAVK: Lokalisation der Einetagenerkrankungen
|
Beckentyp: Aorta/ A. iliaca, fehlende Pulse ab Leiste, Ischämieschmerz Gesäß, Oberschenkel
Oberschenkeltyp: A. femoralis/ A. poplitea, fehlende Pulse ab A. poplitea, Ischämieschmerz Wade Peripherer Typ: Unterschenkel- /Fußarterien, fehlende Fußpulse, Ischämieschmerz Fußsohle |
|
|
Schweregradeinteilung der pAVK
|
nach Fontaine-Ratschow:
Stadium I: Beschwerdefreiheit Stadium II: Claudicatio intermittens (a schmerzfreie Gehstrecke > 200m, b schmerzfreie Gehstrecke < 200m) Stadium III: Ischämischer Ruheschmerz Stadium IV: Nekrose, Gangrän, Ulkus |
|
|
nichtinvasive Diagnose pAVK
|
Knöchel/Arm-Index im CW-Doppler < 0,9 (normal 0,9-1,2)
evtl. Druckmessung nach Belastung (20 Zehenstände): Abfall des Knöcheldrucks unter 90% des Systemdrucks Orientierend mittels Pulsoxy: SO2 an der Großzehe >2% niedriger als am Zeigefinger pathologisch Bei sehr hohen Knöchelarteriendrücken -> Mönckeberg´-Mediasklerose (Diabetes Typ II) |
|
|
Poplitelaes Entrapment
|
Kompression der A. poplitea durch Fehlanlage des M. gastrocnemius oder stark hypertropierte Wadenmuskulatur
-meist einseitig, typisch junge Männer -kann zu Verschluss von Unterschenkelarterien führen -aktive Plantarflexion führt zu Verlust des CW-Doppler-Signals |
|
|
Unterschied zwischen Nekrose und Gangrän
|
Nekrose: schwarz, trocken, Demarkationsgrenze
Gangrän: feucht, infiziert, meist ohne Demarkation |
|
|
Klink der hochgradigen Aortenstenose
|
-eingeschränkte Belastbarkeit
-Dyspnoe -Angina pectoris (auch ohne KHK) -Schwindel, Synkopen |
|
|
OP-Indikationen für Aorteninsuffizienz
|
1. Symptomatische Patienten (ab NYHA II oder Angina pectoris)
2. Asymptomatische Patienten mit EF < 50% 3. Asymptomatische Patienten mit EF > 50% aber LVEDD > 70mm (endsystolisch > 50mm) 4. Unabhängig vom Schweregrad der AI bei Aortendilatation (Marfan >45mm, bikuspide Klappe und A. asc. >50mm, sonstige > 55mm) |
|
|
M. Winiwarter-Buerger
|
Thombangitis obliterans (TAO)
Nicht arteriosklerotische, tabakrauchassoziierte immunmediierte Endarteriitis, sekundäre Thrombosierung des Gefäßlumens Krankheitsbeginn vor dem 40.LJ In Japan bis 16% aller art. Verschlüsse evtl. Fußsohlenclaudicatio, Phlebitis migrans, Nekrosen an Finger- und Zehenkuppen! Typisch: Korkenzieherkollateralen Therapie: Verzicht auch Rauchen, Prostaglandin E, ASS Amputationsrate bis 30% |
|
|
Subclavian Steal Syndrom
|
A. subclavia-Veschluss proximal des A. vertebralis-Abgangs führt zu einer Strömingsumkehr in der A. vertebralis. Bei Armarbeit kommt es zu Schwindel und Sehstörungen. Blutdruckdifferenz zwischen den Armen (> 20mmHg)
|
|
|
Stadien der AVK viszeraler Gefäße
|
I Symptomlos
II Angina abdominalis (postprandiale Schmerzen) III Wechselnde Dauerschmerzen und Malabsorption, evtl. mit ischämischer Kolitis IV Akuter Mesenterialarterienverschluss mit Mesenterialinfarkt (initial heftiger Schmerz, beschwerfreies Intervall, nach Stunden akutes Abdomen, Durchwanderungsperitonitis, Ileus |
|
|
Versorgungsgebiet der A. mes. sup.
|
Flexura duodenojejunalis bis zur linken Kolonflexur
|
|
|
Welche Medikamente können Vasopsasmen im Splanchnicusgebiet auslösen?
|
Digitalis
Ergotamin |
|
|
Bauchaortenaneurysma
|
95% infrarenal, 20% Ausdehnung in die Beckenarterien
Querdurchmesser > 3cm OP-Indikation: Männer >5,5cm, Frauen > 4,5cm |
|
|
Thorakales Aorteneneurysma
|
Aorta ascendens 50%
Aortenbogen 10% Aorta descendens 40% >3,5cm OP-Indikation >5,5cm oder rasche Zunahme OP-Risiko bei Descendensersatz höher |
|
|
Takayasu Arteriitis
|
Granulomatöse Erkrankung der Aorta und ihrer Hauptäste, meist Pat. < 40Jahre
( Vaskulitis der großen Gefäße) ACR-Kriterien (>3): -Alter < 40 Jahre -Claudicatio intermittens der oberen/ unteren Extremitäten -Pulsabschwächung der A. brachialis ("pulseless disease") -Blutdruckdifferenz > 10mmHg -Gefäßgeräusche -pathologisches Angiogramm Sono: konzentrische echoarme Wandverdickung mit Halo (Makkaroni-Phänomen) BSG hoch Therapie: -Immunsuppression -ASS, evtl. Marcumar -evtl. Stenosetherapie |
|
|
Kawasaki-Syndrom
|
Mukokutanes Lymphknotensyndrom
Häufigste Vaskulitis bei Kleinkindern (Vaskulitis mittelgroßer Gefäße) 6 Hauptsymptome (>5): 1. Septische Temperaturen, nicht auf AB ansprechend 2. Doppelseitige Konjunktivitis 3. Stomatitis 4. Palmaerythem, Plantarerythem 5. Exantheme mit an den Fingerspitzen beginnenden Schuppung 6. Schwellung der Halslymphknoten Ko: Aneurysmen der Koronarien Endothelzell-AK (AECA) Th: Hochdosiert Immunglobuline, ASS, kein Kortison! |
|
|
Aortendissektion
|
Intimaeinriss
Stanford A (60%): Aortenbogen, Aorta ascendens Stanford B (40%): Aorta descendens Einteilung in 5 Klassen: 1. Klassische AD 2. Intramurales Hämatom 3. Lokalisierte AD 4. Plaqueruptur mit AD 5. Traumatische AD oder iatrogene AD 2 Prädilektionsstellen: 1. 2-3cm distal der Aortenklappe, 2. am Übergang des beweglichen Aortenbogens in die fixierte Aorta descendens |
|
|
Komplikationen bei Aortendissektion
|
-Blutverlust aus dem falschen Lumen in das paraaortale Gewebe bzw. Ruptur in die freie pleurahöhle
-akute Aorteninsuffizienz mit Überlastung des linken Ventrikels -Akute Perikardtamponade -Schlaganfall -Bild des Gefäßverschlusses einer Extremität -Verlegung der Nieren-/ Mesenterialarterien |
|
|
Raynaud-Syndrom
|
1. Primär (>50% d. Fälle): Durch Kälte oder emotionalen Stress ausgelöste, anfallsartige Vasospasmen der Finger, symmetrisch
Sekundäres Raynaud-S.: Sklerodermie, M. Winiarter-Buerger, Vibrationsschäden, Karpaltunnelsyndrom, pAVK, hämatolog. Erkr. Trikolore: Blässe durch Vasospasmus, Akrozyanose durch Paralyse der Venolen, Hautrötung durch reaktive Vasodilatation Faustschlussprobe, Eiswasserprobe Th: Nifedipin |
|
|
Stadieneinteilung der Varikosis
|
nach Marshall:
I: Keine Beschwerden, allenfalls kosmetisch störend II: Stauungsgefühl, nächtliche Krämpfe III: Ödem, Hautinduration, Pigmentierung, abgeheitltes Ulcus cruris IV: Ulcus cruris venosum |
|
|
Stadieneinteilung der chronisch-venösen Insuffizienz
|
nach Widmer:
I Reversible Ödeme, Corona phlebectatica, perimalleoläre Knötchenvenen II Persistierende Ödeme, Dermatosklerose, Atrophie blanche, Stauungsekzem, zyanotische Hautfarbe III floride oder abgeheilte Ulcera cruris |
|
|
Thrombusformen
|
1. Abscheidungsthrombus (Plättchenthrombus, weißer Thrombus): Anlagerung an der Gefäßwand durch Endotheldefekt, fest an der Gefäßwand heftend, nicht das ganze Lumen ausfüllend
2. Gerinnungsthrombus (roter Thrombus): Strömungsverlangsamung, keine feste Haftung 3. Gemischter Thrombus (weißer Kopf, roter Schwanz) |
|
|
Hereditäre Ursachen einer Thrombophilie
|
1. APC-Resistenz (30%) = Faktor V Leiden Mutation (gestörte Inaktivierung von Faktor Va durch aktiviertes Protein C
2. Prothrombin-Mutation (10%): erhöhter Prothrombinspiegel (F II) 3. Protein C Mangel (5%) 4. Protein S Mangel (2%) 5. AT III Mangel (<1%) |
|
|
Gerinnungskaskade
|
Intrinsic: XII-> XI -> IX -> VIII
-> X Extrinsic: III (tissue factor) -> VII -> X Gemeinsamer Weg: X -> V -> Prothrombinaktivator -> Prothrombin (II) -> Thrombin -> Fibrinogen (I) -> Lösliches Fibrin -> stabiles Fibrin |
|
|
Quick/ INR
|
Thromboplastinzeit, erfasst die Verminderung der Faktoren I, II, V, VII, X; II, VII und X sind Vitamin K-abhängig
|
|
|
PTT
|
Partielle Thromboplastinzeit
Suchtest für Defekte des endogenen Gerinnungssystems (XII, XI, IX, VIII) und der gemeinsamen Endstrecke (X,V, II, I) |
|
|
Protein C und S
|
Vitamin-K-abhängige Faktoren des Gerinnungssystems:
Protein C wird durch Thrombin zu aktiviertem Protein C (APC). APC inaktiviert F Va und VIIIa und befördert die Freisetzung von Gewebe-Plasminogen-Aktivator (t-PA). Die Wirkungen von Protein C werden durch Komplexbildung mit Protein S verstärkt. Mangel führt zu erhöhtem Thromboserisiko |
|
|
Wie hemmen Heparine, Hirudine und Cumarine die Gerinnung?
Dagibatran, Rivaroxaban? |
Heparin: Hemmung von Thrombin indirekt durch Aktivierung von physiologischem Antithrombin
Hirudine (Liperudin - Refludan): Direkte Hemmung von Thrombin Dagibatran (Pradaxa): Direkte Hemmung von Thrombin Fondaparinux (Arixtra): Direkte Hemmung von Faktor Xa Rivaroxaban (Xarelto): Direkte Hemmung von Faktor Xa Cumarine: Vitamin-K-Antagonisten (Vit K = Kofaktor bei der Karboxylierung der Faktoren des Prothrombinkomplexes (II, VII, IX, X) |
|
|
Zeichen einer TVT
|
Trias: Schwellung, Schmerz, Zyanose
Meyer´Zeichen: Wadenkompressionsschmerz Payr´Zeichen: Fußsohlenschmerz bei Druck auf die mediale Fußsohle Homans´Zeichen: Wadenschmerz bei Dorsalflexion des Fußes |
|
|
Phlegmasia coerulea dolens
|
Perakuter Verschluss sämtlicher Venen einer Extremität mit sekundärer Kompression der art. Zirkulation durch rasche Ödembildung
|
|
|
Antiphospholipidsyndrom
|
Diagnosekriterien:
-eine oder multiple Thrombosen (arteriell oder venös); Myokardinfarkte, Hirninsulte, M. Raynaud -Schwangerschaftskompl. -Nachweis von Antiphospholipid-AK (Anti-Cardiolipi-AK, Anto-Beta2-Glykoprotein-AK, Lupuskoagulans) Außerdem: -Thrombopenie -Hämolyse 50% primär, 50% im Rahmen einer Grunderkrankung (Malignome, AIDS, SLE) |
|
|
Paget-von-Schroetter-Syndrom
|
Tiefe Armvenenthrombose
Ursachen: -Halsrippe -Scalenus-anterior-Syndrom -Kostoklavikuläres Syndrom -Hyperabduktionssyndrom (Sehne des M. pectoralis) -Kallus nach Klavikulafraktur Di: Adson-Manöver: Abduktion und Elevation des Arms, dabei Zurücklegen und Drehen zur kontralateralen Seite des Kopfes -> Radialispuls verschwindet |
|
|
Phlegmasia coerulea dolens
|
Perakuter Verschluss sämtlicher Venen einer Extremität mit sekundärer Kompression der art. Zirkulation durch rasche Ödembildung
|
|
|
Antiphospholipidsyndrom
|
Diagnosekriterien:
-eine oder multiple Thrombosen (arteriell oder venös); Myokardinfarkte, Hirninsulte, M. Raynaud -Schwangerschaftskompl. -Nachweis von Antiphospholipid-AK (Anti-Cardiolipi-AK, Anto-Beta2-Glykoprotein-AK, Lupuskoagulans) Außerdem: -Thrombopenie -Hämolyse 50% primär, 50% im Rahmen einer Grunderkrankung (Malignome, AIDS, SLE) |
|
|
Woran muss man denken, wenn unter Heparin Thrombosen auftreten?
|
HIT II -> White Clot Syndrome
Nicht die Heparindosis erhöhen, sondern Heparin absetzen! |
|
|
Ziel INR bei verschiedenen Erkrankungen
|
Mechanische Herzklappen:
Zweiflügelklappen oder Kippklappen -Aorta 2,0-3,0 (2,5-2,5 bei VHF) -Mitralis 2,5-3,5 (+100mg ASS bei VHF) Bioprothesen 2,0-3,0 für 3 Monate Lungenembolie 2,0-3,0 VHF 2,0-3,0 |
|
|
Akuter Arterienverschluss einer Extremität
|
6xP nach Pratt
1. Pain 2. Paleness 3. Paresthesia 4. Pulselessness 5. Paralysis 6. Prostration (Schock) Sofortmaßnahmen: Extremität tief lagern, Volumengabe (Schockprophylaxe), 10.000IE Heparin |
|
|
Lungeninfarkte
|
Treten nur bei 10% aller Lungenembolien auf.
Größere Embolien mit Verlegung mittlerer Pulmonalarterien führen wegen der Ausgleichsversorgung zwischen Bronchial- und Pulmonalarterien nicht zu einem Lungeninfarkt. |
|
|
Vorhersagewahrscheinlichkeit für eine Lungenembolie
|
Wells-Score
-Symptome einer frischen TVT (3) -LE wahrscheinlicher als andere Diagnosen (3) -Hf > 100/min (1,5) -OP oder Immobilisation (1,5) -Frühere TVT oder LE (1,5) -Hämoptysen (1,0) -Tumorerkrankung (1,0) <2: geringe Wahrscheinlichkeit 2-6: mittlere W. >6: hohe W. |
|
|
Dauer der Cumarintherapie bei Thrombembolien
|
Erste Thrombembolie
-transienter Risikofaktor: 3 Monate -idiopathische Genese oder Thrombophilie: 6-12 Monate -kombinierte Thrombophilie oder Antiphospholipid-AK-Syndrom: 12 Monate Rezidivierende Thrombembolie oder Tumorerkrankung: Zeitlich unbegrenzt |
|
|
Stadieneinteilung bei Lymphödem
|
0 Latenzstadim
I Weiche Schwellung II Beginnende Fibrose der Haut III Elephantiasis (stark fibrotisch verdickte, derbe Haut) |
|
|
In welcher Reihenfolge erscheinen bei einem bakteriellen Infekt Leukozyten im Blut?
|
1. Neutrophile Kampfphase (Linksverschiebung, toxische Granulation)
2. Monozytäre Überwindungsphase 3. Lymphozytär-eosinophile Heilphase |
|
|
DD Neutrophilie
|
-bakterielle Infektionen
-Stress (Adrenalin) -Kortison -myeloproliferative Erkrankungen -Raucherleukozytose -Trauma -Gewebsnekrosen (Herzinfarkt) -Kollagenosen -Gichtanfall, thyreotoxische Krise |
|
|
DD Eosinophilie
|
Merkwort PANIC
Parasiten Allergien Neoplasien (auch paran.) Immunologie Cutis (Psoriasis) |
|
|
Bei welchen bakteriellen Infektionskrankheiten findet man normale oder verminderte Granulozyten?
|
Typhus
Brucellose |
|
|
Brucellose
|
-gramnegative Stäbchen mit intrazellulärer Vermehrung
-Weltweite Anthropozoonose, betr. bes. Landwirte, Schäfer, Metzger, Tierärzte -B. melitensis (Schafe und Ziegen); B. abortus (Rinder), B. suis (Schweine), B. canis (Hunde) 90% subklinisch Epitheloidzellige, nicht verkäsende Granulome im RHS (LK, Milz, Leber) und Gefäßwänden -Kontakinfektionen oder perorale Infektion durch Milchprodukte Kl.: Fieber, unregelmäßiger Verlauf, Hepatosplenomegalie, LK-Schwellungen, Kopf-, Muskel-, Gelenkbeschwerden, im Verlauf Organmanifestationen: Granulome in Leber, Milz, Knochen - Brucella-Arthritis Ko.: Endokarditis, Osteomyelitis, Meningoenzephalitis Verlauf akut, subakut oder chronisch, setzen sich im RHS fest; geringe Letalität Th.: Doxycyclin + Streptomycin |
|
|
Leptospirose
|
-Leptospira interrogans, >200 Serovare (z.B. L. icterohaemorrhagica -> M. Weil)
Weltweise Zoonose, Ratten, Mäuse, Nagetiere; gefährdet sind Angler, Wassersportler, Kanal- und Feldarbeiter Übertragung durch Urin (Schleimhäute, Aerosole) Biphasischer Verlauf (M. Weil): 1. Sepsis: Brutaler Beginn mit hohem Fieber, Konjunktivitis, Exantheme, Kopfschmerzen 2. Organmanifestationen: ikterische Hepatitis, Nephritis, Meningitis, Enzephalitis Ko.: Nierenversagen, Leberversagen Di.: Erreger PCR aus Blut, Liquor (1. Wo), Urin (ab 2. Wo), AK (ab 2. Wo) Th.: Peni G oder Ceph 3 Letalität bis > 20% |
|
|
Typhus abdominalis
|
Salmonella typhi
bes. verbreitet in Indien, Pakistan, Indonesien Übertragung von Mensch zu Mensch, ab ano ad os, wichtigste Infektionsquelle sind Dauerausscheider, indirekte Inf. durch Trinkwasser oder kont. Lebensmittel -Langsamer Beginn mit langsam steigenden Temp., septische Fieberkontinua ohne Schüttelfrost, Bradykardie -Splenomegalie -Roseolen der Bauchhaut -Kopfschmerzen, Husten -Typhuszunge (graugelb belegt, rote Ränder) -Benommenheit (Typhos = Nebel) -Anfangs Obstipation, später erbsbreiartiger Durchfall -Leukopenie Ko.: Meningitis, Hämatochezie, Myokarditis, Nierenversagen Dauerausscheider (10 Wo nach Beginn noch Salmonellen im Stuhl), Gallenblase oder Dünndarm Th.: Ciprofloxacin oder Ceph. 3 Oraler Lebendimpfstoff, parenteraler Totimpfstoff |
|
|
Granulozytopenie
|
Bildungs- /Reifungsstörungen im Knochenmark:
-Chemikalien (Benzol) -Medikamente -Strahlen -Knochenmarksinfiltration -Osteomyelosklerose -Vit B12 oder Folsäuremangel Gesteigerter Umsatz: -Verbrauch bei bkt. Inf. -Verteilungsstörung (Hyperspelnismus) -Immunneutropenien |
|
|
Agranulozytose
|
Medikamentös induzierte Immungranulozytopenie mit plötzlicher Zerstörung aller Granulozyten
Auslöser: -Metamizol -NSAR -Carbimazol, Thiamazol -Sulfonamide, Sulfasalazin, Cotrimoxazol -Clozapin, Clomipramin -Rituximab (CD 20 AK) Medikament (Hapten) + Plasmaprotein verbinden sich zum Vollantigen -> bei wiederholter Zufuhr AK, Leukozytolyse Trias: Fieber, Angina tonsillaris, Stomatitis aphthosa |
|
|
Häufige primäre Immundefekte
|
-Selektiver IgA-Mangel
-Spezifischer AK-Mangel bei normalen Immunglobulinen -CVID (common variable immunodeficiency): 2 Altersgipfel, late onset hypogammaglobulinemia -SCID (Schwere kombinierte Immundefekte) Weitere: -Di George Syndrom (CATCH - cardiac defect, abnormal face, thymic hypoplasia, cleft palate, hypocalcemia) -Louis Bar Syndrom (Ataxia teleagiectasia) -Wiskott Aldrich Syndrom (Thrombozytopenie) |
|
|
DD Lymphozytose
|
-Virusinfekte
-lymphozytäre Heilphase bakterieller Infekte -Tbc, Lues, Pertussis, M. Bang -Methadon Substitution -CLL |
|
|
DD cervicale Lymphknoten
|
-EBV
-Toxoplasmose -Röteln -HIV -M. Hodgkin, NHL -HNO-Tumoren -SD-Karzinom |
|
|
5 exanthematische Infektionskrankheiten
|
1. Scharlach (feinfleckig, Mund-Kinn-Dreieck frei)
2. Röteln (makulopapulös, mittelfleckig, nicht konfluierend) 3. Ringelröteln (gilrandenförmig makulopapulös, periodisches Abblassen und Neuentstehen) 4. Masern (grobfleckig, konfluierend) 5. Windpocken (Sternenhimmel) |
|
|
Scharlach
|
Beta-hämolysierende Streptokokken der Lancefieldgruppe A
pyrogene Exotoxine A und C, Immunität nur gegen die Exotoxine Kl.: Angina tons., Husten, hohes Fieber, submand. LK, ab 4.Tag Himbeerzunge, ab 2. Tag Exanthem (stecknadelkopfgroß, Beginn Achseln, Leisten, periorale Blässe, im Verlauf kleieförmige Hautschuppung) Rumpel-Leede-Test pos. Ko.: -Toxischer Verlauf mit Myokarditis, Erbrechen, Durchfall, Benommenheit -Sinusitis, Otitis media -Septischer Verlauf, Meningitis, Hirnsinusthrombose -Streptokokkenallergische Nacherkrankungen: Rheumatisches Fieber, Glomerulonephritis 10 Tage Penicillin oder Makrolid |
|
|
Röteln
|
Rubivirus (RNA-Virus)
Kl.: Bei Kindern in 50% d. Fälle asymptomatischer Verlauf Leichter Beginn, mittelfleckige, nicht konfluierendes makulopapulöses Exanthem, Beginn hinter den Ohren, LK im Nackenbereich Ko.: Enzephalitis, Purpura durch passagere Thrombopenie, Arthritis Embryopathie: Auge, Ohr, Herz, geist. Retard. La.: Leukopenie, Lymphozytose |
|
|
Ringelröteln
|
Parvovirus B 19
-> Ziel sind die erythropoetischen Zellen im KM Kl.: -Ringförmiges oder girlandenförmiges makolupapulöses Exanthem, livide Wangenverfärbung, periodisches Abblassen und Neuentstehen des Exanthems -Anämie, Thrombopenie, Granulopenie Ko.: Bei Pat. mit chron. hämolyt. Anämie -> hämolytische Krise; Schwangerschaft Hydrops fetalis |
|
|
Masern
|
Paramyxovirus (RNA-Virus)
Infektiös 5 Tage vor bis 4 Tage nach Auftreten des Exanthems Kl.: 1. Prodomi: Rhinitis, Konjunktivitis, Fieber, bellender Husten, Enanthem, Koplik´sche Flecken Wangenschleimhaut 2. Exanthem: Großfleckig, konfluierend, Beginn hinter den Ohren; Hals-LK- Schwellung Ko.: -Otitis media -Pneumonie (virale Masernpneumonie, bkt. Superionfektion, Risenzellpneumonie) -Laryngotracheitis mit Krupp -Enzephalitis (akut postinfektiös, subakute sklerosierende Panenzephalitis - SSPE) -selten: foudroyanter Verlauf |
|
|
Varizella-Zoster-Virus-Infektionen
|
1. Windpocken: Fieber, polymorphes Bild der Hauterscheinungen (Sternhimmel): Roseolen, Papeln, Bläschen, Krusten. Ko: Selten Enzephalitis; Pneumonie mit hoher Letalität
2. Zoster: Reaktivierung der VZV, die nach der Primärinfektion in den Spinalganglien persistieren. Begünstigend: Stress, Sonne, Immunschwäche, Malignom. Häufigste Lok.: Thorakal, ein oder mehrere Dermatome, Ko: Postzosterische Neuralgien, Zoster ophthalmicus, Zoster oticus mit Fascialis Parese, Meningoenzephalitis Th.: Aciclovir 3x10mg/kg i.v. oder 5x800mg p.o. |
|
|
Mononukleose
|
EBV, Zielzellen: naso- und oropharyngealen Epithelien und B-Lymphozyten
"Kissing disease" Kl.: Trias: Fieberhafte Angina tonsillaris, LK-Schwellungen, "buntes" Blutbild mit Virozyten -Milzschwellung, cave Ruptur -Exanthem, Arzneimittelexanthem durch Gabe von Ampicillin oder Amoxicillin -Hepatitis -selten Panzytopenie -Meningoenzephalitis, Myokarditis -TINU-Syndrom (tubulo-interstitielle Nephritis + Uveitis) -chronischer Verlauf mit anhaltender Virusreplikation EBV-assoziierte Malignome: -Posttransplatations-lymphoproliferative Erkrankungen -B-Zell-Lymphome bei AIDS -Nasopharynxkarzinom -Burkitt-Lymphom -orale Haarleukoplakie Labor: Virozyten (mononukleäre Zellen), Paul-Bunnel-Reaktion, AK |
|
|
Cytomegalie
|
CMV (DNA-Virus)
Inf.: Schmier- und Tröpfchen-Infektion, Bluttransfusion, Organtransplantation, sexuell Interstitielle lymphoplasmazelluläre Entzündung mit Riesenzellen und viralen Einschlusskörperchen (Eulenaugenzellen) Kl.: -konnatale Infektion: Frühgeburt, Hydrozephalus, zerebrale Verkalkungen, Chorioretinitis, Hepatosplenomegalie, Hörschäden, Intelligenzdefekt -Immunkompetente Erwachsene > 90% der Erkrankungen asymptomatisch, evtl. mononukleoseähnliches Krankheitsbild -Immunsupprimierte Erwachsene: Mononukleoseähnliches Krankheitsbild, Retinitis bei AIDS (Cotton-Wool-Herde), Enzephalitis, interstitielle Pneumonie, Hepatitis, GI-Manifest., Leukopenie, Thrombopenie Th.: Ganciclovir, Valganciclovir |
|
|
Dreitagefieber
|
HHV 6
Plötzliches hohes Fieber, 3-5 Tage lang, mit Fieberabfall Auftreten eines blassrötlichen Exanthems, häufig Fieberkrämpfe, Kleinkinder |
|
|
ABVD-Schema
|
Behandlung des M. Hodgkin:
Adriamycin Bleomycin Vinblastin Dacarbazin Wdh. am Tag 29 |
|
|
BEACOPP-Schema
|
Behandlung des M. Hodgkin
Bleomycin Etoposid Adriamycin Cyclophosphamid Oncovin=Vincristin Procarbazin Prednison Wdh. am Tag 22 |
|
|
M. Hodgkin
|
Monoklonales B-Zell-Lymphom
Mehrkernige Sternberg-Riesenzellen und einkernige Hodgkinzellen in den Keimzentren der LK, buntes Bild durch Bystander-Zellen 2 Altersgipfel (30. LJ, 60. LJ) Kl.: -LK-Befall 60% Kopf-Hals-Gebiet, 95% oberhalb des Zwerchfells; Kartoffelsack (zu Paketen verbackene LK) -evtl. Hepatosplenomegalie -B-Symptome: Fieber (Pel-Ebstein-Fieber, häufig bei abdom. Manifest.), Nachtschweiß, Gewichtsverlust > 10% KG in 6 Monaten -Leistungsminderung -evtl. Juckreiz -LK-Schmerzen nach Alkoholgenuss Prognose: 90% aller Rezidive in den ersten 5 Jahren, 5-Jahres-Überleben: Limitierte St. >90%, intermed. St. 90%, fortgeschr. St. bis 88%; aber: Toxizität der Therapie (erhöhtes Risiko für Zweitmalignome, Kardiotox., Lungentox.) |
|
|
erhöhtes Risiko für M. Hodgkin
|
-HIV
-EBV -immunsuppressive Therapien -Holzschutzmittel |
|
|
Histologische Klassifikation des M. Hodgkin
|
A Klassisches Hodgkin-Lymphom (93%)
1. Noduläre Sklerose (60%) 2. Mischtyp (28%) 3. Lymphozytenreicher Typ (5%) 4. Lymphozytenarmer Typ (0,3%) B Lymphozyten-prädominantes Hodgkin-Lymphom (7%) |
|
|
Stadienteinteilung des M. Hodgkin/ NHL
|
Ann Arbor Klassifikation
I Befall einer LK-Region/ 1 extranod. Herd II 2 oder mehr LK-Regionen auf einer Seite des Zwerchfells III 2 oder mehr LK-Regionen beiderseits des Zwerchfells III1 oberhalb des Tr. coeliacus III2 unterhalb des Tr. coeliacus IV Disseminierter Befall extralymph. Organe mit oder ohne LK-Befall Zusatz: A ohne Allgemeinerscheinungen B B-Symptomatik |
|
|
Therapie des M. Hodgkin
|
Limited Disease: IA - IIB ohne RF: 2x ABVD + 30Gy IF-RT
Intermediär: IA - IIB mit RF (hohe BSG/ >= 3 LK-Areale): 2x BEACOPP + 2x ABVD + 30 Gy IF-RT Advanced disease: IIB mit RF (zusätzlich extranod. Befall/ großer Mediastinaltumor), III, IV: 8x BEACOPPP + RT, Pat. > 60J. 8x ABVD Rezidiv: -Reinduktionstherapie mit anschließender Hochdosischemoth. und autolog. Stammzelltransplantation |
|
|
Langzeittoxizität der Radio-/Chemotherapie beim M. Hodgkin
|
-erhöhtes Risiko für Zweitneoplasien
-Kardiotoxizität durch Anthrazykline und RT -pulmonale Toxizität durch Bleomycin und RT -Infertilität -SD-Funktionsstörungen |
|
|
Ätiologie Myokarditis
|
1. infektiös:
-50% Viren (Parvo B 19, Coxsackie B1-B5, A, HHV6, EBV, Influenza, Adeno, Echo, HIV, HCV -Bkt. bei Sepsis -Beta-Hämol.- Strept. -Borrelia Burgdorferi (Lyme-Disease) -Diphtherie -Toxoplasmose, Chagas -Trichinen, Echinokokken 2. nicht-infektiös: -RA, Kollagenosen, Vaskulitiden -RT -idiopathische Fiedler-Myokarditis |
|
|
Chagas-Krankheit
|
Häufigste Ursache einer inflammatorischen DCM in Südamerika
Trypanosoma cruzi (Kot von Raubwanzen) Kl.: Akut: lokale schmerzlose Schwellung an der Eintrittstellen (Chagom),; Fieber, LK-, Leber-, Milzvergr. Chronisch: Trias: DCM, Megaösophagus, Megakolon Th.: Nifurtimox, Benznidazol (nur im Akutstadium möglich) |
|
|
Hon-Hodgkin-Lymphome
|
1. Niedrig maligne NHL
2. Hochmaligne NHL AIDS-Pat. haben eine bis 1000-fach erhöhte Inzidenz WHO-Klassifikation: NHL der B-Zell-Reihe (z.B. Diffuses großzelliges B-Zell-Lymphom - DLBCL (30%; hochmaligne), B-CLL, Follikuläre Lymphome (25%), Mantelzell-Lymphom, Plasmozytom, Burkitt-Lymphom) und der T-Zell-Reihe (z.B. T-CLL, Mycosis fungoides) Ät.: -Immundefekte (Immunsuppressiva, Zytostatika, AIDS, Autoimmunerkrankungen) -Exposition gegenüber Radioaktivität -Viren (HTLV, EBV), Helicobacter (MALT-L.) -Gifte (Benzol, Toluol) |
|
|
MALT-Lymphom
|
Mucosa associated lymphatic tissue
90% der niedrig malignen MALT-Lymphome sind Folge einer chronischen H.p.-Besiedlung, Eradikation kann zur Heilung führen. |
|
|
R-CHOP
|
Rituximab
Cyclophosphamid Doxorubicin Vincristin Prednison Behandlung einiger NHL |
|
|
Multiples Myelom
|
Plasmozytom, M. Kahler
B-Zell-NHL mit diffuser oder multilokulärer Infiltration des KM, Klon maligne transformierter Plasmazellen -> monoklonale Immunglobuline IgG (54%), IgA (25%), IgD (1%), Leichtketten (20%) Ossermannkriterien (2/3): 1. Monoklonale Immunglobuline 2. Plasmazellnester im KM 3. Osteolytische Herde/ Osteoporose (Schrotschussschädel) B-Symptomatik Lab.: Extrem hohe BSG, Bence-Jones-Proteine, M-Gradient in der E´phorese, Anämie, Hyperkalzämie Ko: -Spontanfx -Myelomniere (tox. Effekt der Leichtketten auf Nierentubuli) -nephrotisches Syndrom -Hyperkalzämische Krise -Hyperviskositätssyndrom -Polyneuropathie -selten Plasmazellleukämie, ZweitTu, AML -AL-Amyloidose Verlauf: progredient (90%) oder smoldering DD MGUS (monoklonale Gammopathie unbestimmter Signifikanz) |
|
|
Häufigster Tumor von KM und Knochen
|
Plasmozytom
|
|
|
Stadieneinteilung des Plasmozytom
|
nach Durie and Salmon:
I Hb>10g/dl, Ca normal, Rö normal/ max 1 Osteolyse, IgG < 5g/dl, IgA < 3g/dl, LK < 4g/24h II weder I noch III III Hb <8,5g/dl, Ca erhöht, Knochenveränderungen, IgG > 7g/dl, IgA > 5g/dl, LK > 12g/24h A Krea < 2mg/dl B Krea > 2mg/dl Weitere Einteilung nach Höhe des B2M (<3,5mg/l; 3,5-5,5; >5,5) |
|
|
MGUS
|
Monoklonale Gammopathie unbestimmter Signifikanz
3% aller Pat. >70J -konstant niedrige Konz. des monoklonalen Immunglobulins -<10% klonale Plasmazellen im KM -keine Osteolysen Risiko für die Entwicklung eines MM 1%/Jahr |
|
|
Therapie des Plasmozytoms
|
Indikation: Endorganschäden (CRAB-Kriterien) oder Hyperviskiositätssyndrom, gehäufte bakterielle Infekte
CRAB-Kriterien: Hyperkalzämie (C) Niereninsuffizienz (R) Anämie (A) Knochenbeteiligung (B) Pat. < 70J: Induktionstherapie, danach Hochdosis-Mephalantherapie mit autologer Stammzelltransplantation, evtl. 2. Transplantation, Erhaltungstherapie mit Lenalidomid Pat. > 70J: MPT (Mephalan, Prednison, Thalidomid) oder MPV (Bortezomib = Velcade) Thalidomid und Lenalidomid = antiangiogenetisch Bortezomib: Protease-Inhibitor Rezidiv: 2. Transplant., evtl. Auto/allo Konzept Außerdem: -Bisphosphonate -lokale RT -Impfungen gegen Pneumok., Influenza, HiB -Erypo, GCSF, IgG |
|
|
Prognose bei Plasmozytom
|
St. I 64 Monate
St. II 32 Monate St. III bis 12 Monate |
|
|
Immunozytom
|
M. Waldenström, Makroglobulinämie
B-Zell-NHL mit Bildung monoklonaler IgM-Globuline 4x seltener als Plasmozytom, höheres Lebensalter, bessere Prognose -keine Osteolysen, nur Osteoporose -keine Hyperkazämie -weniger Nierenschädigung (trotzdem Cave KM-Gabe) -hämorrhagische Diathese durch Bindung von Gerinnungsfaktoren und Aggregation von Thrombozyten -AIHA Hyperviskositätssyndrom (Raynaud, Sehstörungen) palliative Therapie, Rituximab |
|
|
Prognostischer Index beim Immunozytom
|
Alter (65J)
4 Risikofaktoren: 1. Hb<11,5g/dl 2. Thrombozyten < 100.000/yl 3. B2M > 3mg/l 4. IgM > 70g/l 5 Jahres ÜL: 36-87 Monate |
|
|
Haarzellleukämie
|
niedrig malignes B-NHL
Panzytopenie durch Markfibrose -> Infekte; Splenomegalie Chemotherapie beim Auftreten von Symptomen (Purinanaloga, Interferon A, Rituximab beim Rezidiv) |
|
|
CLL
|
Leukämisch verlaufendes B-Zell-NHL, niedrig maligne; häufigste Leukämieform, höheres Alter
Kl.: -LK-Schwellungen (immer) -evtl. Splenomegalie -Hauterscheinungen (Pruritus, Urtikaria, Herpes zoster u. simplex, Mykosen, knotige Infiltrate) -Parotisschwellung und Tränendrüsenbefall (Mikulicz-Syndrom) Ko.: -AK-Mangel -> Infekte -AIHA -Hypersplenismus -zelluläres Hypervsikositätssyndrom -Transformation in eine sekundär hochmalignes NHL (Richter Syndrom) La.: -permanente Lymphozytose, Gumprecht-Kernschatten -B2M, AK-Mangel, monokl. Immunglob. |
|
|
DD Pruritus
|
-Allergie
-Darmparasiten -CLL und andere maligne Lymphome -Polycytämia vera -Eisenmangel -Diabetes mellitus -Niereninsuffizienz -seniler Pruritus -Cholestase, PBC, PSC |
|
|
CLL Stadieneinteilung
|
nach Binet
A < 3 vergr. LK-Regionen (ÜLZ >10J) B >= 3 vergr. LK-Regionen (ÜLZ 5-7Jahre) C Hb < 10g/dl, Thrombopenie < 100.000/yl, LK-Status irrelevant (ÜLZ < 3J) oder RAI-Klassifikation 0 nur Lymphozytose I +LK-Vergr. II + Hepato- /Splenomegalie III + Anämie IV + Thrombopenie |
|
|
Therapie CLL
|
Symptomatische Patienten im St. B, alle Pat. im Stadt. C
Guter AZ: FCR (Fludarabin, Cyclophosphamid, Rituximab) Red. AZ: Chlorambucil (Leukeran) oder Bendamustin; ggf. +Rituximab Rezidiv: Alemtuzumab, Ofatumumab Ggf. RT großer Lymphome, Kortison bei AIHA oder Thrombopenie, Impfung gegen Pneumokokken und Influenza, ggf. Substitution von Immunglobulinen |
|
|
Monoklonale B-Zell-Lymphozytose ungewisser Signifikanz
|
MBL
<5000 B-Lymphozyten/yl |
|
|
Diagnose der CLL
|
Kriterien des International Workshop on CLL (IWCLL) 2008:
-Nachweis von mind. 5000/yl B-Lymphozyten, Klonalität durchflusszytometrisch gesichert -Vorherrschen von kleinen reifen Lymphozyten -Koexpression von CD19, CD20 und CD23 und CD5 KM-Biopsie ist oft nicht notwendig. |
|
|
Lymphome der B-Zell-Reihe
|
KM:
Stammzelle Pro-B-Zelle Prä-B-Zelle -> lymphoblastisches Lymphom vom B-Zell-Typ Peripehres lymph. Gewebe: Reife naive B-Zelle: B-CLL, Mantelzell-Lymphom Keimzentrumszelle: Burkitt-Lymphom, Follinkuläres Lymphom, diffuses großzelliges B-Zell-Lymphom, M. Hodgkin Memory-B-Zellen: Maginalzonenlymphom Plasmazelle: Plasmozytom |
|
|
Primäre und sekundäre lymphatische Gewebe
|
Primär:
Knochenmark (B-Zell-Bildung) Thymus (T-Zell-Bildung) Senkundär: Milz LK MALT (Mucosa-assoziiertes lymphatisches Gewebe) |
|
|
Gastrointestinale Lymphome
|
bleiben lange lokalisiert
80% B-Zell-Lymphome 10% T-Zell-Lymphome (Sprue) Magenschleimhaut hat normalerweise kein MALT, Akquisition von gastralem MALT durch chron. H.p.-Gastritis Endoskopie: NHL entstehen primär in der Submukosa, es kommt erst sekundär zu einer Wandinfiltration Bei niedrig malignen MALT-Lymphomen des Magens durch H.p.-Eradikation in 70-90% der Fälle Vollremission |
|
|
Kutane T-Zell-Lymphome
|
1. Mycosis fungoides:
-Prämykosides Stadium: wie Ekzem -Infiltratives Stadium -Tumorstadium mit Befall von Milz, LK, anderer Organe 2. Sézary Syndrom: Trias: Generalisierter Hautbefall, LK-Schwellungen, leukämisches Blutbild mit Sézary-Zellen |
|
|
ALL
|
80% im Kindesalter
Ungünstige Prognose bei: -Leuko>30.000/yl -Alter > 50J -Zytogenetic: t(9;22), t(4;11) -Sub-Typ: pro-B-ALL -Zeit bis zur Remission > 4 Wochen |
|
|
AML
|
80% im Erwachsenenalter
Ungünstige Prognose: -Leuko > 100.000/yl -Alter > 60J -Therapiekurse bis Remission > 1 -Zytogenetik: abn (3q), 5/5q, 7/7q, abn (12p), abn(17p), komplex veränderte Karyotypen 3 Hauptgruppen: AML ohne chromosomale Aberrationen (Heilungsrate bis 30%) und mit balancierten chromosomalen Aberrationen (HR >60%) und mit unbalancierten Aberrationen (HR <15%) |
|
|
Klinik der akuten Leukämien
|
1. Allgemeinsymptome: Fieber, Nachtschweiß, Abgeschlagenheit
2. Verdrängung der normalen Hämatopoese -Granulozytopenie -> Schleimhautenzündungen, bkt. Infekte -Anämie -> Blässe, Dyspnoe, Müdigkeit -Thrombozytopenie -> Blutungen; DIC 3. Weitere Symptome -LK-Schwellungen (30%), Splenomegalie -Meningeosis leucämica (bes. ALL) -Hypertrophische Gingivitis (bes. AML) -Haut- und Organinfiltration -Knochenschmerzen bei kindl. ALL |
|
|
Labor bei akuten Leukämien
|
-Leukozyten könnten normal, erhöht oder vermindert sein.
-Unreife Zellen in Blut und KM -Anämie, Thrombopenie, Granulopenie (Hiatus leucaemicus) -BSG erhöht -evtl. LDH und Harnsäure erhöht -Myeloblasten-Leukämie: Auer-Stäbchen |
|
|
Ätiologie der Leukämien
|
-Benzol, Zytostatika
-Ionisierende Strahlen -HTLV1 Viren (Südjapan, Karibik) -Trisomie 21, Klinefelter Syndrom -MDS, aplastische Anämie, Osteomyelofibrose |
|
|
Therapie der Leukämien
|
Remissionsinduktion -> Konsolidierung (-> Reinduktion) -> Remissionserhaltung
ALL: Prednison, Vincristin, Daunorubicin, L-Asparaginase; Methotrexat intrathekal, RT des Schädels; Remissionserhaltung mit Methotrexat, 6-Mercaptopurin AML: ARA-C (Cytarabin), Daunorubicin Allogene Transplantation von hämatopoetischen Stammzellen nach Konditionierung (intensive Chemotherapie + Ganzkörperradiatio) |
|
|
Akute und chronische GvHD bei Leukämien nach SZT
|
Akut: innerhalb von 100 Tagen nach SZT
Schädigung von 1. Haut 2. Darm (Durchfälle) 3. Leber (Hepatitis) Chronisch: -Sicca Syndrom -papulöses Exanthem -Schleimhaut wie erosiver Lichen ruber |
|
|
Philadelphia Chromosom
|
Vorkommen bei 85% aller CML (20% ALL des Erwachsenen, 5% ALL Kinder)
Reziproke Translokation t(9;22)(q34;q11). Das Chromosom 22 (Philadelphia Chromosom) zeigt dadurch ein Fusionsgen bcr-abl-> Kodierung eines Fusionsproteins mit Tyrosinkinase-Aktivität (proliferationsfördernd, Apoptose-hemmend) |
|
|
CML
|
Klassische CML: Philadelphia Chromosom (85%)
3 Krankheitsphasen: 1. Chronisch stabile Phase (4-6 Jahre) 2. Akzelarationsphase (1Jahr) 3. Blastenkrise Typisch sind Splenomegalie und sehr hohe Leukozytenzahlen! Durch hohe Leukozytenzahlen leukämische Thromben (Milzinfarkte, Zentralvenenthrombosen der Retina, leukämischer Priapismus, Myokardinfarkte) Labor: -Leukozytose durch Vermehrung der neutroph. Granulozyten, Linksverschiebung, Basophilie; Anämie, Thrombozytose, kernhaltige Vorstufen Pseudo-Gaucher-Zellen im KM prognostische günstig |
|
|
extramedulläre Blutbildung
|
Blutbildung außerhalb des KM, in der Fetalzeit physiologisch
Leber, Milz, Haut Vorkommen: CML, Osteomyelofibrose Typisch sind kernhaltige rote Vorstufen im Blut. |
|
|
Therapie der CML
|
Imatinib (Glivec): Tyrosinkinase-Inhibitor führt zur kompletten hämatologischen Remission in ca. 95% der Fälle, 400mg/d Dauertherapie
Bei Auftreten von Resistenzen neue Tyrosinkinaseinhibitoren, ggf. allogene KMT oder SZT (einzige Möglichkeit der Heilung) |
|
|
chronische myeloproliferative Erkrankungen
|
-Polycythaemia vera
-Essentielle Thrombozythämie -Osteomyelofibrose |
|
|
Polycythaemia vera
|
Häufigste Ursache: Mutation des JAK2-Gens
Gesteigerte Produktion aller 3 Zellreihen, wobei die Erythropoese im Vordergrund steht. Kl.: -Plethora (Rötung Gesicht) und Rötung Extremitäten -aquagener Pruritus -Erythromelalgie (plötzliche schmerzhafte Rötung und Überwärmung der Füße) -Schwindel, Kopfschmerzen, Ohrensausen -Splenomegalie Ko.: -Thrombembolie -Hämorrhagische Diathese -Entwicklung eines MDS oder einer akuten Leukämie (15%/20J) -KM-Insuffizienz Th.: -Aderlässe -ASS -ggf. Hydroxyurea (Litalir) (erhöhtes Risiko für die Induktion einer späteren akuten Leukämie) |
|
|
DD Erythrozytose
|
1. Stresserythrozytose
2. Hämokonzentration durch Exsikkose 3. EPO-Vermehrung (paraneoplasisch, sekundär durch Hypoxie oder Hämoglobinstörungen; exogene Zufuhr) 4. Hormonale Stimulation (M. Cushing, Therapie mit Kortison oder Androgenen) 5. Polyzythaemia vera |
|
|
Essentielle Thrombozytämie
|
Monoklonale autonome Proliferation der Thrombozytose
Kl.: -Thrombozyten > 1.000.000/yl -Mikrozirkulationsstörungen -Thrombembolie -Hämorrhagische Diathese durch funktionsgestörte Thrombos durch vermehrte Bildung des vWF -Splenomegalie WHO-Diagnosekriterien: A1 Thrombo dauerhaft > 600.000/yl A2 KM mit vergrößerten reifen Megakaryozyten A3 JAK2-Mutation B1 Ausschluss von PV, CML, OMF, MDS Th.: Hydroxyurea + ASS bei Hochrisikopat. (kein ASS bei Thrombo > 1.000.000/yl), oder Anagrelid->Hemmung der Mekgakaryozyten und Hemmung der Thrombozytenaggregation, oder Alpha-Interferon; bei intermdiärem Risiko ASS Transformation in akute Leukämie in 10% der Fälle |
|
|
Osteomyelofibrose
|
Trias:
1. Hochgradige Markfibrose 2. Extramedulläre Blutbildung 3. Milzvergrößerung BB: A Hyperproliferative Frühphase (Leukozytose, Thrombozytose) B Spätphase mit Panzytopenie, weiße und rote Vorstufen im Blut durch extramedulläre Blutblidung, Poikilozytose "Trockenes Mark": Puncio sicca Th: -Alpha-Interferon zur Senkung der Thrombzyten -Thamidomid, ggf. Kombination mit Prednisolon Mittlere Überlebenszeit 5 Jahre, 10% der Pat. entwickeln ein MDS oder eine myeloische Leukämie |
|
|
Aplastische Anämie
|
Eine exogene Noxe (Virus, Medikament, Benzol) führt bei genetischer Disposition zu einer Autoimmunreaktion gegen hämatopoetisches Gewebe.
Kl.: Panzytopenie (Manchmal auch Mono- oder Bizytopenie) Aplastisches Knochenmark Th.: -SZT (KMT oder PBSCT) -Immunsuppressiva -Substitution der Zellen, supportive Therapie |
|
|
Meylodyplastische Syndrome
|
Heterogene Gruppe erworbener klonaler KM-Erkrankungen (viele verschiedene Subtypen)
Periphere Zytopenie, zellreiches KM Häufig Transformation in eine AML Therapie anhand von Prognose-Scores: Niedrigrisiko-MDS: supportiv, Wachsttumsfaktoren der Hämatopoese, Immunsuppressive Therapie, Lenalidomid Hochrisiko-MDS: Azacytidin; Polychemotherapie, allogene SZT |
|
|
Stufentherapie Antiemetika unter Chemotherapie
|
1. Leicht: Dexamthason
2. Mittel: Setron (z.B. Odansetron=Zofran) + Dexa 3. schwer: Aprepitant (Neurokinin-1-Rezeptorantagonist) + Setron + Dexa |
|
|
Karnofsky-Index
|
100% Normale Aktivität
90% gering verminderte Belastbarkeit 80% deutlich verminderte Belastbarkeit 70% Keine normale Aktivität, Pat. versorgt sich selbst 60% gelegentliche Hilfe erforderlich 50% ständige Pflege und häufige ärztl. Hilfe erforderlich 40% überwiegend bettlägrig 30% dauernd bettlägrig 20% schwerkrank 10% moribund |
|
|
ECOG-Skala
|
Grad 0: Normal aktiv
Grad 1: Mäßig eingeschränkt, eingeschränkt arbeitsfähig Grad 2: arbeitsunfähig, >50% pflegebedürftig Grad 3: >50% kontinuierliche Pflege Grad 4: 100% bettlägrig |
|
|
WHO-Stufenschema Schmerztherapie
|
Stufe 1: Nicht-Opiodanalgetika
Stufe 2: schwaches Opioid + nicht Opioidanalgetikum Stufe 3: starkes Opioid + Nicht-Opioidanalgetikum Bedarfsmedikation 10-20% der Gesamttagesdosis |
|
|
Amyloidose
|
Störung der Proteinfaltung -> Ablagerung von Proteinen als unlösliche fibrilläre Aggregate in Organen, Gefäßen und Nerven
Nachweis: blau nach Kontakt mit Jod, Eosinophilie in HE-Färbung, Doppelbrechung im polarisierten Licht, Beta-Faltblatt (Rö-Beugung) Betroffene Organe: "speckartig" (Speckleber, Speckmilz) 1. Systemische Amyloidosen: -Typ AA (chronisch inf./entzünd. Erkr.; fam. Mittelmeerfieber, Malignome) -Typ AL (monoklonale Gammopathien) -ATTR (erblich) 2. Lokalisierte Amyloidosen: -Diabetes mellitus, medulläres SD-Ca, M. Alzheimer, seniles kard. Amyloid betroffene Organe: -Nieren (nephrot. Syndrom) -Herz (restriktive Kardiomyopathie) -peripheres Nervensystem -Makroglossie -Hepato-/Splenomegalie (Speckleber, Speckmilz) -Shoulder Pad Sign -periorbitale Blutungen (Bindung von Fk. X) |
|
|
Familiäres Mittelmeerfieber
|
familiär rekurrente Polyserositis
autosomal rezessiv vererbt, im Mittelmeerraum verbreitet Kl.: Schübe mit hohem Fieber und Schmerzen der Serosa (Peritoneum, Pleura, Gelenkkapseln). Dauer 2-3 Tage. 90% der Pat. haben ihren ersten Schub vor dem 20.LJ. Ko.: Häufig Amyloidose! Di: Gendiagnostik, Abgrenzung zu anderen periodischen Fiebersyndromen Colchicin zur Prophylaxe, im Schub NSAR |
|
|
Lysosomale Speicherkrankheiten
|
ca. 50 verschiedene bekannt
Bei 2 Erkrankungen ist eine Enzymersatztherapie möglich: 1. M. Gaucher 2. M. Fabry |
|
|
M. Gaucher
|
autosomal rezessiv vererbter Mangel an Glukozerebrosidase (MMS) -> Akkumulation von Glukozerebrosid in den betroffenen Zellen
Viszerale Form: Knochenbeschwerden, Hepato- Splenomegalie, gestörte Hämatopoese Neuropathische Form (akut oder chronisch) Enzymersatztherapie: Rekombinante Glukozerebrosidase |
|
|
M. Fabry
|
X-chromosomal vererbter Mangel an Alpha-Galaktosidase A -> Akkumulation von Globotriaosylceramid (Gb3) im Endothel kleiner Gefäße
-Akroparästhesien -Angiokeratome -Cornea verticulata -Tortuosis vasorum -GI-Beschwerden -Kardiomyopathie -prog. Nierenbeteiligung -Tinnitus und Hörverlust -TIA, Apoplex Enzymersatztherapie |
|
|
Aus welchen Gefäßen besteht der Truncus coeliacus?
|
A. hepatica propria
A. gastrica sinistra A. lienalis |
|
|
Normalgrößen für
V.cava Vv. hepaticae V. porta Aorta |
V.cava 20mm
Vv. hepaticae (Lebervenenstern) 10mm V. porta 11-13mm Aorta 20-25mm |
|
|
Welche Gefäße bilden den venösen Konfluenz?
|
Zuflüsse der V. porta:
V. mesenterica sup. V. mesenterica inf. V. coronaria ventriculi V. lienalis |
|
|
DD Splenomegalie
|
-Pfortaderhochdruck
-Hämolytische Anämie -Myeloproliferative Erkrankungen, Leukosen, Lymphome -Speicherkrankheiten -Infektionskrankheiten (EBV, HIV, Toxoplasmose) |
|
|
Hyperspleniesyndrom
|
Trias:
1. Splenomegalie 2. Zytopenie 3. Knochenmarkshyperplasie Pooling der Blutzellen in der Milz mit vermehrte Sequestration. Di.: 51Cr markierte Erythrozyten (Radiochrom) |
|
|
Folgen einer Asplenie
|
nach Splenektomie oder funktioneller Asplenie bei Sichelzellanämie, nach RT oder bei SLE
1. postoperative Thrombozytose und Lymphozytose 2. Auftreten von Howell-Jolly-Körperchen 3. Verminderte Bildung von IgG und IgM 4. Fehlende Filterfunktion für Bakterien (bes. Kapsel-tragende Bakterien) 5. Verminderte Funktion des MMS OPSI: Overwhelming postsplenectoy infection: Meist akute Pneumokokkensepsis mit DIC und hoher Letalität |
|
|
Sonographisch auffällige pathologsiche Strukturen der Milz
|
-großnoduläre Läsionen bei hochmalignem NHL, diffuse Lymphominfeltration bei niedrigmalignem NHL
-echoreicher Herd: DD Hämangiom -Milzinfarkt: hyporeflexiv, keilförmig -Zysten, Pseudozysten (posttraumatisch) -Milzabszess -Milzruptur |
|
|
Verschiedene Blutungstypen
|
1. Koagulopathie: Hämarthros, Hämatome
2. Thrombozytäre oder vaskuläre hämorrhatgische Diathese: Petechien Purpura (Exanthem aus Petechien) Ekchymosen (kleinflächige Hautblutungen) |
|
|
Blutgerinnung
|
1. Primäre Blutstillung: Vasokonstriktion und Bildung des weißen Plättchenthrombus (ASS verhindert die Bildung von Thromboxan A2 über Hemmung der Cylooxigenase)
2. Sekundäre Blutstillung: Aktivierung der plasmatischen Gerinnungskaskade (Prothrombinaktivator -> Prothrombin ->Thrombin -> Fibrinogen ->lösl. Fibrin -> stabiles Fibrin (+Fk XIII = fibrinstabilisierdender Faktor) |
|
|
AT III Mangel
|
AT III führt zur Inhibition von Thrombin und F. Xa
Ursachen: -verminderte Synthese (Leberzirrhose) -erhöhter Verbrauch (DIC) -erhöhter Verlust (nephrotisches Snydrom, exsudative Enteropathie) |
|
|
Protein C und S
|
Vit. K abhängige Inhibitoren des Gerinnungssystems
Protein C wird durch Thrombin zu aktiviertem Protein C (APC). APC inaktiviert F. Va und VIIIa und fördert die Freisetzung von Gewebe-Plasminogen-Aktivator (t-PA). Verstärkung der Wirkung durch Protein S. Mangel -> Thrombophilie |
|
|
Heparin
|
Indirekte Hemmung von Thrombin durch Aktivierung von physiologischen Antithrombin (bei AT III Mangel ist die Heparinwirkung vermindert.)
|
|
|
Lysetherapie
|
Streptokinase: Aktivierung zusammen mit Plasminogen von Plasmin
tPA: tissue-type plasminogen activator: aktiviert an Fibrin gebundenes Plasminogen (lokale Lyse) |
|
|
Angeborene Koagulopathien
|
1. von-Willebrand-Syndrom (häufigstes)
2. Hämophilie A und B |
|
|
Hämophilie
|
A: Mangel oder Inaktivität von F. VIII
B: Mangel oder Inaktivität von F. IX 50% der Fälle werden X-chromosomal rezessiv vererbt, 50% sporadische Mutationen Kl.: -großflächige Blutungen -Muskelblutungen -Gelenkblutungen, bes. Kniegelenk Di: PTT verlängert, INR normal! Th.: -Leichte Fälle: Desmopressin Nasenspray (bewirkt die Freisetzung der im Endothel gespeicherten Faktoren VIII und vWF) -Schwere Fälle: Substitution von Gerinnungsfaktoren |
|
|
Heyde Syndrom
|
Assoziation von Aortenklappenstenose und v. Willebrand-Syndrom
|
|
|
Gerinnungskasakade merk
|
12-11-9-8 -> 10 (intrinsic, PTT)
3-7 -> 10 (extrinsic, INR) 10-5-2-1 |
|
|
von Willebrand-Jürgens-Syndrom
|
angeboren oder erworben (monoklonale Gammopathie, MPS, autoimmunologische Erkrankungen, Valproisäure)
3 Funktionen des vWF: 1. Verbindung der Thrombozyten mit dem Kollagen des Subendothels 2. Thrombozytenaggregation 3. Komplex mit F- VIII -> verzögerter Abbau Kl.: Kombination von hämophilem und petechialem Blutungstyp, Schleimhautblutungen Th.: Desmopressin -> Freisetzung von vWF |
|
|
DIC und Verbrauchskoagulopathie
|
Intravale Aktivierung des Gerinnungssystems führt zu Bildung von disseminierten Mikrothromben in der Endstrombahn -> Verbrauch von Gerinnungsfaktoren
Kl.: -Hämorrhagische Diathese, Blutungen, evtl .Schock -Multiple Fibrin- und Thrombozytenthromben -> Mikrozirkulationsstörung -> akutes Nierenversagen, ARDS, zerebrale Dysfunktion, Leberversagen (MOV) Ursachen: 1. Einschwemmung von Prothrombinaktivatoren in die Blutbahn (Geburtshilfl. Kompl., Hämolysen, Operationen an thrombokinasereichen Organen (Pankreas, Pulmo, Prostata, Plazenta) 2. Indirekte Aktivierung der Gerinnung über Mediatoren (Sepsis, insbes. Meningokokkensepsis) 3. Kontaktaktivierung (extrakorporaler Kreislauf, Mikrozirkulationsstörung im Schock) Man unterscheidet akute DIC und chronische DIC (z.B. bei Malignomen). Labor: Thrombopenie, PTT verlängert, INR erhöht, AT III vermindert, Fibrinogen vermindert, Fibrinmonomere + Fibrinopeptide A positiv (Beweis der intravasalen Gerinnung), D-Dimere erhöht bei sek. Hyperfibrinolyse |
|
|
Tranexamsäure
|
Antifibrinolytikum
(darf nicht bei DIC gegeben werden) |
|
|
Normwerte BB
|
Hämoglobin: M 13-17g/dl, F 12-16g/dl
Hkt: M 42-50%, F 38-44% MCV: 85-98fl MCH: 28-34pg MCHC: 31-37g/dl Reti: 3-18/1000 Ery Leuko: 3.800-10.500/yl Thrombo: 140.000-345.000/yl Neutro: 30-80% Eosino: 0-6% Baso: 0-2% Lympho: 15-50% Mono: 1-12% CD4-Zellen: 435-1.600/yl CD4/CD8: 0,6-2,8 |
|
|
Normwerte Eisenstoffwechsel, Folsäure, Vit B12
|
Eisen M 50-160yg/dl, F50-150yg/dl
S-Ferritin 7-142yg/dl Transferrin-Sättigung 16-45% sTfR 0,83-1,76mg/l Folsäure 7-36nmol/l Vit B12 150-800pmol/l |
|
|
Normwerte E´lyte und Nierenwerte
|
Na 135-145mmol/l
K 3,6-4,8mmol/l Ca 2,2-2,6mmol/l (ionisiert 1,1-1,3) P 0,84-1,45 mmol/l Harnstoff 12-50mg/dl Krea M bis1,1 F bis 0,9mg/dl |
|
|
Normwerte Lipide, Glukose, Harnsäure
|
TG <150mg/dl
Chol <200mg/dl LDL <160 <130 <100mg/dl HDL >40mg/dl Glu nüchtern >= 126mg/dl D.m. OGTT (75g Glukose, 2h) <140mg/dl normal 140-199mg/dl IGT >=200 Diabetes mellitus Harnsäure M bis7,0 F bis 6,0 mg/dl |
|
|
NT-proBNP
|
N-terminale pro brain natriuretic peptide
Auslösender Reiz: Dehnung der Vorhöfe Wirkung: vasodilatatorisch, natriuretisch-diuretisch <400pg/ml HI unwahrscheinlich 400-2000pg/ml HI möglich >2000pg/ml HI wahrscheinlich |
|
|
HELLP-Syndrom
|
Hämolyse
Elevated Liver enzymes Low Platelet count Komplikation der Eklampsie (Hypertonie, Ödeme, Eiweiß im Urin) |
|
|
Helicobacter pylori - Welche Erkrankungen sind assoziiert?
|
1. Ulcus ventriculi, duodeni
2. chron. aktive Typ-B-Gastritis 3. Magenkarzinom 4. MALT-Lymphome 5. M. Werlhof |
|
|
direkter und indirekter Coombs-Test
|
Direkt: Nachweis von IgG, die an Erythrozyten haften (AIHA)
Indirekt: Nachweis von IgG gegen Erythrozyten, die noch frei im zu untersuchenden Serum vorhanden sind (z.B. Rh-Inkompatibilität) |
|
|
Hämoglobinkonstellation beim Erwachsenen
Veränderung bei Thalassämie |
97% HbA (aa/bb)
2,5% HbA2 (aa/dd) <0,5% HbF (aa/gg) Bei der ß-Thalassämie: Minorform -> v.a. HbA2 erhöht Majorform -> v.a. HbF erhöht |
|
|
Häufigste Infektionskrankheiten der Welt
|
1. Tbc
2. Malaria |
|
|
Wie lange kann nach einer Tropenreise noch eine Malaria auftreten?
|
Bis zu 2 Jahre (M. tropica)
90% aller importierten Malariafälle treten innerhalb des 1. Monats nach Rückkehr aus den Tropen auf. |
|
|
Malariaformen
|
1. Benigne Form (typischer Fieberrhyhtmus)
a) Malaria quartana (Plasmodium malariae) immer 2 fieberfreie Tage b) Malaria tertiana (Pl. vivax und ovale) 1 fieberfreier Tag 2. Maligne Form: Malaria tropica (Pl. falciparum) -> unregelmäßiger Fieberrhyhtmus durch fehlende Synchronisation der Parasitenvermehrung |
|
|
Zyklus von Plasmodien
|
Exoerythrozytäre Phase = Leberzyklus
Erythrozytäre Phase = Schizogonie: Morula ->Merozoiten (werden durch Platzen der Erythrozyten frei und verursachen das Fieber) ->Gametozyten ->Mücke (im Magen geschlechtliche Vermehrung, Sporozoiten in der Speicheldrüse) Im Menschen nur ungeschlechtliche Vermehrung. |
|
|
Klinik der Malaria
|
-Fieber (bei M. quartana - P. malariae - und tertiana - P. vivax/ ovale - typischer Fieberrhythmus - 48/72h, bei M. tropica - P. falciparum - unregelmäßiges Fieber, mögl. subfebril)
-Kopf- und Gliederschmerzen -Schmerzen im rechten Oberbauch, Hepato-/Splenomegalie -Übelkeit, Erbrechen, Durchfall -Hämolytische Anämie, Thrombozytopenie! Hypoglykämie Bei M. tropica: Mikrozirkulationsstörungen mit Ischämie wichtiger Organe (zerebral, Herz, Lunge, Nieren) -> Krampfanfälle, Nierenversagen, Lungenödem, DIC, Schock Bei Malaria tertiana und quartana Rezidive möglich (2,5,50J), bei Malaria tropica keine Rezidive. |
|
|
Diagnose der Malaria
|
Dicker Tropfen, 2xtägl. an 2 aufeinanderfolgenden Tagen, 30 Minuten lang mikroskopieren
Molekularbiologische Diagnostik Nachweis von Plasmodien-AK |
|
|
Therapie der Malaria
|
Benigne Form:
Cloroquin, im Anschluss Primaquin bei Pl. vivax und ovale, da keine Wirkung auf extraerythrozytäre Formen) Maligne Form (M. tropica) unkompl. Verlauf: Atovaquon + Proguanil Mefloquin Artemether + Lumefantrin kompl. Verlauf: Chinin + Doxycyclin (wegen Chinin-Resistenzen) evtl. Austauschtransfusion Cave Hypoglykämie Cave Lungenödem |
|
|
Cloroquin (Resochin)
|
Therapie der benignen Malariaformen und Prophylaxe
Cave: Bei langfristiger Einnahme Retinaschädigung möglich |
|
|
Mefloquin (Lariam)
|
Prophylaxe und Therapie der unkomplizierten Malaria tropica, notfallmäßige Selbstbehandlung
Cave: Nebenwirkungen! (GI, neurolo, psychiatr., kardial, hämatol.) |
|
|
Gelbfieber
|
RNA-Virus
Übertragung durch Stechmücken und evtl. Zecken Gelbfiebergürtel: Tropisches Afrika, tropisches Süd-/Mittelamerika Kl.: 1. Initialstadium (3 Tage, plötzliches hohes Fieber, Kopf- und Muskelschmerzen, Konjunktivitis, rel. Bradykardie) 2. Remissionsstadium Fieberabfall mit evtl. Ausheilung 3. Stadium der hepatorenalen Schädigung (wenn keine Ausheilung): Hepatitis mit Ikterus, Nephritis, hämorrhagische Diathese Ko.: Leber-/Nierenversagen, Meningoenzephalitis, Multiorganversagen, Koma Bis zum 14. LJ gute Prognose, bei Erwachsenen bis 85% Letalität. Schutzimpfung |
|
|
Dengue-Fieber
|
Flavi-Virus, Übertragung durch Aedes aegypti und Aedes albopictus
Asien, Südamerika, USA, Karibik >90% der Infektionen verlaufen asymptomatisch oder wie grippaler Infekt 1. Stadium: Plötzlicher Krankheitsbeginn mit hohem Fieber und Knochen- und Muskelschmerzen ("breakbone fever"), Kopfschmerzen, metallischer Geschmack im Mund 2. Stadium: Nach vorherigem Fieberabfall erneuter Fieberanstieg, Exanthem, das an Masern erinnert, LK-Schwellungen Besonders bei Kindern in Endemiegebieten oft schwerwiegender Verlauf. Bei 2% thrombozytopenische Blutungsneigung -> hämorrhagischer Schock. Zweiinfektion gravierender als Erstinfektion. |
|
|
Bilharziose
|
Schistosomiasis; Tropen, Subtropen
S. haematobium (Blase) S. mansoni, S. japonicum (Mesenterialgefäße) Gabelschwanz-Zerkarien aus Süßwasserschnecken (Zwischenwirt) penetrieren die Haut (Zerkariendermatitis). Wanderung über Lymphgefäße, rechtes Herz in die Lunge (evtl. Katayama-Syndrom mit Fieber, Husten, Eosinophilie) -> Zerkarien gelangen zur Leber und entwickeln sich dort zu Würmern. Blasenbilharziose: Hämaturie Darmbilharziose: Kolitis -erhöhtes Risiko für Blasenkarzinom bzw. Kolonkarzinom -portale Hypertension (Verstopfung der Pfortader durch Eier) Th.: Praziquantel (Heilungsrate > 80%) |
|
|
Leishmaniose
|
Protozoen
L. donovani-Komplex (L-infantum, L.chagasi) -> viszerale L. L. tropica und L. peruviana -> kutane L. (Orientbeule) L. brasiliensis -> mukokutane L. Übertragung durch Phlebotome (Schmetterlingsmücken, sandflies), Reservoir: Nagetiere Nach dem Stich Wanderung in LK, Milz, Leber, KM Lebenslang Reaktivierung möglich. Viszerale L.: Trias Hepato-Splenomegalie, Panzytopenie, Gammaglobuline erhöht, Kala Azar (fleckartige dunkle Pigmentierung der Haut) Kutane L.: Orientbeule Mukokutane L.: Zusätzlich Schleimhautveränderungen an Nase und Rhinopharynx, evtl. mit ausgedehnten Zerstörungen Th.: Liposomales Ampho-B; kutane L. Lokalbehandlung |
|
|
Problemkeime in Kliniken
|
MRSA (Methicillin-resistenter Staphylococcus aureus)
VRE (Vancomycin-resistente Enterokokken) ESBL (Extended Spectrum Beta-Lactamasen) z.B. bei E.coli und Klebsiellen KPC (Klebsiella-pneumoniae Carbapenemasen) |
|
|
CAP bei jungen und alten Erwachsenen; häufige Erreger
|
Junge Patienten:
-Pneumokokken (30-50%) -Haemophilus influenzae (bis 10%) -Chlamydia pneumoniae (bis 10%) -Legionellen -Mykoplasmen -Pneumotrope Viren als Bahnung für bkt. Superinfektionen (Influenza A und B, Adenovirus, Parainfluenza, SARS-Coronavirus) Pat. > 65J: wie bei jungen Patienten, zusätzlich gramnegative Bakterien (Klebsiellen, Enterobacter, E. coli) |
|
|
HAP: Häufige Erreger
|
(ab 5. Tag der Hospitalisierung):
Am häufigsten gramnegative Bakterien (Pseudomonas, Enterobacter, E.coli, Proteus, Serratia, Klebsiella pneumoniae) Beatmungsassoziierte Pneumonie: S. aureus, P. aeruginosa, Klebsiellen, Enterobacter, E.coli |
|
|
Pneumonie-Erreger bei Pat. mit herabgesetztem Immunstatus
|
-Pneumoncystis jiroveci
-Pilze -Viren (Zytomegalie, Herpes simplex, Varizella zoster) -atypische Mykobakterien |
|
|
Stadien der Lobärpneumonie
|
1. Anschoppung (1.Tag): dunkelrote, blutreiche Lunge, Crepitatio indux
2. Rote Hepatisation (2./3. Tag): Fibrinreiches Exsudat führt zu leberartiger Konsistenz der grauroten Lunge 3. Graugelbe Hepatisation (4.-8.Tag): Leukozyteninfiltration 4. Lysis (nach dem 8. Tag): Enzymatische Verflüssigung des Fibrins, Leukozytenzerfall, Abhusten des eitrigen Auswurfs; Auskultation: Crepitatio redux (Alveolen wieder lufthaltig) |
|
|
Pathologisch-anatomisch Einteilung der Pneumonien
|
1. Lobärpneumonie (z.B. Pneumokokken)
2. Lobuläre Pneumonie (=Herdpneumoinie) (Pneumokokken, Streptokokken, Staphylokokken), häufigste Form = Bronchopneumonie 3. Akute interstitielle Pneumonie (Viren, Mykoplasmen, Rickettsien, Chlamydien) 4. Miliarpneumonie (Tbc, Histoplasmose, Coccidiomykose) |
|
|
Klinik der akuten Pneumonie
|
Lobärpneumonie:
-Schüttelfrost, hohes Fieber -Husten, Dyspnoe, Nasenflügeln -begl. Herpes labialis -Thoraxschmerzen durch Begleitpleuritis, bei diaphragmaler Beteiligung Fortleitung des Schmerzes in den rechten Oberbauch -Rotbraunes Sputum ab 2. Tag -Auskult: Bronchialatmen, pos. Bronchophonie, klingender RGs, pos. Stimmfremitus) -Am 7.-9. Tag Entfieberung mit evtl. lebensbedrohlihcer Herz-Kreislauf-Belastung (prä AB-Ära) Atypische Pneumonie (Chlamydien, Mykoplasmen, Legionellen, Viren): -Beginn langsam -Cephalgien, Myalgien, nur leichtes Fieber -trockener Reizhusten mit spärlichem/ fehlendem Auswurf -Missverhältnis zwischen geringem Auskultationsbefund und pos. Rötngenbefund. -normale Leukos, evtl. Lymphozytose |
|
|
Rumpel-Leede-Zeichen
|
Blutdruckmanschette >diastol, <systol.
-> Nach 10 Minuten Stauung Petechien distal der Manschette |
|
|
Akromegalie
|
Hyperpituitarismus
Somatotropes Adenom des HVL mit Überproduktion von GH (Growth hormone) Regelkreis: Die Sekretion von GH wird durch GHRH (GH-Releasing hormone) angeregt und durch Somatostatin gebremst. Wirkung von GH über IGF-1 (insulin like growth factor 1 = Somatomedin C). Durch Hyperglykämie Suppression der GH-Ausschüttung. Kl.: Bis zum Erwachsenenalter: Gigantismus -Veränderung der Physiognomie (Vergröberung der Gesichtszüge, Vorstehendes Kinn, Verlängerung der Nase, Vorspringen der Augenbrauen) -Verlängerung von Zehen und Fingern (Schuhe, Handschuhe, Hüte passen nicht mehr) -Vergrößerung der Zunge, kloßige Sprache -Vergrößerung der inneren Organe (Splanchmomegalie, Kardiomegalie) -Sehstörungen (bitemporale Hemianopsie) -path. Glukosetoleranz -Karpaltunnelsyndrom -vermehrtes Auftreten von Mamma- und Kolonkarzinomen Th.: transsphenoidale Adenotomie, Strahlentherapie, Dopaminantagonisten (Bromocriptin), GH-Rezeptorantagonisten (Pegvisomant), STH-Analoga (Octreotid) |
|
|
Perkussion der Lungengrenzen
|
Lungen-Leber-Grenze: ICR 6/7
Skapularlinien: 9.Rippe Paravertebral: 11.BWK Atem-Verschieblichkeit normal ca. 6 cm |
|
|
Rasselgeräusche der Lunge
|
1. Trocken: bronchitisch durch Schleimfäden oder Schleimmembranen in den Bronchien
->Brummen (niederfrequent), Giemen (hochfrequent) 2. Feucht: Bronchus mit dünnflüssgiem Sekret gefüllt (Ödem, Blut, Eiter), wie Strohhalm in Wasserglas feinblasig-> Erkrankungen der Endverzweigungen der Bronchien grobblasig -> Erkrankungen der größeren Äste Klingend -> ohrnah (Leitfähigkeit der Lungengewebes für hohe Frequenzen durch Infiltration verbessert) 3. Knisterrasseln -Entfaltungsknistern -Fibroseknistern, Sklerosiphonie 4. Pleurareiben: Lederknarren, Schneeballknirschen (in- und exspiratorisch) |
|
|
Endokarditisprophylaxe -Wer?
|
1. Pat. mit Herzklappenersatz (mechanisch und biologisch)
2. Pat. mit rekonstruierten Klappen unter Verwendung von alloprothetischem Material in den ersten 6 Monaten nach OP 3. Pat. mit überstandener Endokarditis 4. Pat. mit angeborenen zyanotischen Herzfehlern und operierte Pat. mit Conduits; operativ oder interventionell behandelte Herzfehler unter Verwendung von alloprothetischem Material in den ersten 6 Monaten nach OP -Pat. nach Herztransplantation mit kardialer Valvulopathie |
|
|
Endokarditisprophylaxe - Wann?
|
-Zahnbehandlungen
-Eingriffe am Respirationstrakt (Adenotomoi, TE, starre Bronchoskopie) -Eingriffe am GIT, RT und UGT nur noch bei bestehenden Infektionen |
|
|
Endokarditisprophylaxe - Was?
|
Einzeldosis 30-60min vor Eingriff
Amoxicillin 2g p.o. oder Ampicillin 2g i.v. Bei Penicillinallergie: Clindamycin 600mg p.o. oder i.v. oder Ceftriaxon 2g i.v. oder Cefalexin 2g p.o. oder Clarithromycin 500mg p.o. |
|
|
Nichtinfektiöse Endokarditis
|
1. E. rheumatica
2. Libman-Sacks-Endokarditis (SLE) 3. Löffler-Syndrom (Endokarditis eosinophilica) im Rahmen verschiedener Erkrankungen (z.B. Asthma bronchiale, Periarteriitis nodosa, Hodgkin-Lymphom, NHL, Lungenkarzinom, eosinophile Leukämie) |
|
|
Häufigste Erreger der Endokarditis
|
45-65% Staphylokokken
30% Streptokokken <10% Enterokokken, gram-negative Bakterien Nativklappenendokarditis: Methicillinsensible Staphylokokken, Strptokokken, Enterococcus faecalis Frühe Endokarditis nach Klappenersatz: MRSA, koagulase-negative Staphylokokken, gram-negative Erreger |
|
|
Klinik der Endokarditis
|
1. Fieber, Allgemeinsymptome
2. Kardiale Symptome: Herzgeräusch, Herzinsuffizienz, Rhyhtmusstörungen 3. Kutan: Petechien, Splinter-Blutungen unter den Nägeln, Osler-Knötchen, Janeway-Läsionen 4. Bakterielle Mikroembolien (embolische Herdenzephalitis, Niereninfarkte) 5. Nierenbeteiligung (glomeruläre Herdnephritis - Löhlein) 6. Splenomegalie 7. Augen: Roth´s-Spots (Retinablutungen) Verlaufsformen: 1. Akute Sepsis (typisch für Staphylokokken) 2. Endokarditis lenta (typisch für Viridans Streptokokken) -> schleichender Verlauf mit unklarem Fieber |
|
|
Kalkulierte Initialtherapie der Endokarditis bei unbekanntem Erreger
|
Nativklappen:
Ampicillin i.v. + Gentamycin (evtl. nur 2 Wochen) + Cefotaxim oder Ceftriaxo Dauer: 4-6 Wochen Bei fehlendem Ansprechen evtl. Carbapenem bzw. Vanco + Genta Klappenprothesen: Vancomycin + Gentamycin + Rifampicin Dauer: Genta 2 Wochen, beide andere >= 6 Wochen |
|
|
Dringliche OP-Indikationen bei inf. Endokarditis
|
-persistierende Infektion
-AV-Blockierungen -paravalvulärer Abszess -Herzinsuffizienz -hämodynamisch relevantes Klappenvitium -Embolien -Vegetationen >10mm |
|
|
Synkope
|
1. Neurokardiogene Synkope (Reflexsynkope):
Verminderung der Sympathicus- und Zunahme der Parasympathicusaktivität -> RR-Abfall und Bradykardie (vasodepressorisch, kardioinhibitorisch, Mischtyp). ->Karotissinus-Syndrom, vasovagal, pressorische Synkopen durch Husten, Miktion. Typisch sind Prodromi. 2. Orthostatische Synkope: Plötzliches Aufstehen aus liegender Position oder längeres Stehen -> Versagen des vasokonstriktorischen Reflexes im Bereich der Kapazitätsgefäße (Venen der Beine) (sympathikotone OH, asympathikotone OH, POTS (posturales orthostatisches Tachykardiesyndrom) -> autonome Neuropathie (z.B. M. Parkinson, Diabetes), Volumenmangel 3. Kardiale Snykope Arrhythmie, strukturelle Herzerkankungen (z.B. HOCM, Aortenstenose) |
|
|
DD Art. Hypotonie
|
-Primär, kein Krankheitswert
-medikemtös -endokrin (Hypothyreose, NNR-Insuffizienz, HVL-Insuffizienz, Hypoaldosteronismus) -kardiovaskulär (Aortenstenose, Herzinsuff., Rhythmusstörungen, pulm. HTP, konstriktive Perikarditis) -Immobilisation -Hypovolämie, Hyponatriämie |
|
|
Carotisdruckversuch
|
-Durchführung empfohlen bei Pat. > 40 Jahre mit unklarer Synkope
-KI bei Pat. mit TIA oder Stroke < 3 Monate oder Strömungsgeräuschen über der A. carotis -Diagnose hypersensitiver Caortissinus wenn Asystolie >= 3 s oder RR-Abfall um 50mmHg bei Synkope |
|
|
Stehversuch
|
Diagnose OH:
Symptomatik + Abfall RR systol. >= 20mmHg, RR diast. >= 10mmHg oder Abfall RR systol. < 90mmHg Ohne Symptomatik Diagnose erwägen. |
|
|
Kipptischuntersuchung
|
15 Minuten liegend, max. 45 Minuten 60-70°
Indikation: Dient zum Nachweis von Reflexsynkopen -Erste Synkope bei High-Risk-Patienten (berufl. Konsequenzen) -Wiederholte Synkopen Diagnose NCS (VVS): Induktion von Hypotension und Bradykardie + Synkope Diagnose erwägen bei fehlenden Symptomen |
|
|
LZ-EKG: Diagnose Synkope
|
-Zeitliche Korrelation zwischen Synkope und brady- oder tachykarder Rhythmusstörung
-AV-Block Mobitz II oder III -Ventrikuläre Asystolie >= 3 Sekunden -prolonged SVT oder VT (>32 Schläge mit Hf>160/min) |
|
|
Pneumonieerreger
|
-Pneumokokken
-Hämophilus influenzae -Mykoplasmen -Legionellen -Chlamydien -Coxiella burnetti -Adenoviren, Influenza, SARS-Corona -Pneumocystis -Candida, Aspergillose, Kryptokokkose |
|
|
Pneumokokken
|
Streptococcus pneumoniae, grampositive Diplokokken, >40 Serogruppen, >90 versch. Kapsel-Polysaccharide
Infektion meist endogen -Otitis media, Sinusitis -Meningitis, Pneumonie, Sepsis Pat. nach Splenektomie besonders gefährdet für invasive Verläufe (OPSI). Th.: Aminopenicillin + Beta-Lactamase-Inhibitor (z.B. Amoxycillin + Calvulansäure); Cephalosporine III Polyvalenter Polysaccharidimpfstoff |
|
|
Haemophilus influenzae
|
-Nasopharyngitis
-Epiglottitis bei Kleinkindern -Pneumonie bei Kleinkindern und Erwachsenen mit chronischen Lungenerkrankungen -Meningitis bei Kleinkindern Th.: Erwachsene Chinolone Kinder Cephalosporine III Aktive Immunisierung möglich |
|
|
Mykoplasmen
|
Mykoplasma pneumoniae: Kleinste frei vermehrbare Lebewesen ohne feste Zellwand
10-20% asymptomatisch 80% Tracheobronchitis 5-10% interstitielle Pneumonie Ko.: -Bakterielle Superinfektion -AIHA durch Kälteagglutinine Nachweis von DNA, AK Th.: Makrolide oder Doxycyclin |
|
|
Influenza A
|
Subtypenklassifizierung anhand von 2 in die Virushülle eingebauten Glykoproteinen:
H: Hämagglutinin (Anheften an die Wirtszellen) N: Neuraminidase (Freisetzung von Viren aus infizierten Zellen) Antigendrift: Kleine Antigenveränderungen durch Punktmutationen -> Epidemien alle 2-3 Jahre Antigenshift: Reassortment, es werden ganze Genabschnitte ausgetauscht (in Schweinen zwischen humanen und aviären I.-Viren) ->Pandemien 1918/19 A(H1N1) (spanische Grippe) 1957/58 A(H2N2) (asiatische Grippe) 1968/69 A(H3N2) (Hongkong-Grippe) 2009 A(H1N1/09) Schweine-Grippe, Mexiko-Grippe |
|
|
Vogelgrippe
|
A(H5N1): Nur für Vögel hoch ansteckend, Übertragung von Vögeln auf Menschen sporadisch, keine Ansteckung von Mensch zu Mensch
50% Letalität! Eine humane Pandemie könnte durch Reassortment von H5N1 mit einem humanen Influenza-Virus ausgelöst werden. |
|
|
Klinik und Therapie der Influenza A
|
Kl.:
80% asymptomatisch oder leichte Erkältungsbeschwerden -plötzlich hohes Fieber, eingipfelig -starkes Krankheitsgefühl -Laryngo-Tracheobronchitis mit trockenem Husten -Rhinitis, Pharyngitis -Kopf-, Glieder- und Muskelschmerzen Ko.: -Pneumonie --Primär hämorrhagische Influenzapneumonie --interstitielle Grippepneumonie --sekundär bakterielle Grippepneumonie -Purpura Schoenlein Hennoch -Myoperikarditis Verzögerte Rekonvaleszenz Th.: innerhalb der ersten 48 Stunden Zanamivir (inhalativ) Oseltamivir (Tamiflu) Peramivir (i.v. USA) |
|
|
Staphylokokkenpneumonie
|
1. aerogen (meist Superinfektion auf dem Boden einer primären Virusinfektion): Herdförmige peribronchiale Infiltrate
2. hämatogen (Folge einer Staphylokokkensepsis, ausgehend von Hautläsionen oder septischen Thrombophlebitiden: Disseminierte Herde Häufig septisches Krankheitsbild, hohe Letalität Rö.: -herdfömige Infiltrate -Einschmelzungen und ggf. Höhlenbildung -häufig bronchiolitische Streuung -häufig Pleurabeteiligung -Entwicklung von Pneumatozelen |
|
|
Legionellose
|
Legionellen, L - pneumoniae: Gramnegative, intrazellulär wachsende aerobe Bakterien.
Infektion durch Inhalation infizierter Aerosole aus Wasseranlagen (Whirlpool, etc.), keine Ansteckung von Mensch zu Mensch. Risikopat. = Ältere Menschn, Raucher, Alkoholiker, Pat. mit chron. Erkrankungen. Nur 1% der exponierten Personen erkrankt. 90% Pontiac Fieber (Grippeähnliche Erkrankung ohne Pneumonie) 10% Legionella-Pneumonie: Atypische Pneumonie Di.: Nachweis von Legionella-AG aus dem Urin (Erregernachweis oder DNA aus Bronchialsekret) Th.: Makrolide, Fluorchininolone III und IV, 3 Wochen |
|
|
Chlamydien
|
Chlamydien: obligat intrazellulär lebende Erreger
Chlamydia pneumoniae: Pneumonie, aerogene Tröpfcheninfektion von Mensch zu Mensch, meist leichter Verlauf mit Pharyngitis/ Laryngitis, schwere Verläufe bei älteren Pat./ abwehrgeschwächten Pat. Chlamydia psittaci: Ornithose, Papageienkrankheit; aerogene Infektion durch Kot- und Federstaub von Papageien, Wellensittichen, Tauben, Enten; grippeartiger oder pneumonischer Verlauf; Ko.: Endo-, Myo-, Perikarditis, selten Enzephaltitis Th.: Doxycyclin oder Makrolide für 3 Wochen Chlamydia trachomatis: -Trachom (SG A-C) -Urethritis (SG D-K) -Lymphgranuloma venereum (SG L1-L3) |
|
|
Urethritis
|
Isolierte Harnweginfektion des Pars anterior urethrae distal vom Sphincter urethrae
-Chlamydia trachomatis (häufig; symptomlose Chlamydienträger bis 7% der Männer, bis 20% der Frauen) -Ureaplasma urealyticum -Mykoplasma genitalium -Trichomonas vaginalis -Herpes Typ II -E. coli -Gonorrhö Kl.: Jucken, Brennen, Schmerzen beim Wasserlassen; evtl. Harnröhrenausfluss ("Bonjour-Tröpfchen") Ko.: -Männer: Entzündung von Samenblase und Prostata -Frauen: PID -Perihepatitis nach Gonokokken- oder Chlamydieninfektion (Fitz Hugh Curtis Syndrom) -Reiter Syndrom (Trias Arthritis, Konjunktivitis, Urethritis) |
|
|
Hantavirus-Infektion
|
Hantavirus (RNA-Virus), Erregerreservoir sind Ratten und Mäuse; Infektion durch Einatmen von virushaltigen Ausscheidungen dieser Tiere; verschiedene Serotypen:
1. Hantaan, Seoul, Dobrava: Hämorrhagisches Fieber mit renalem Syndrom (HFRS); weltweit; plötzlicher Beginn mit hohem Fieber, Cephalgien, Myalgien, evtl. Konjunktivitis; Lumbalgien, Abdominalschmerzen, Übelkeit, Erbrechen, Durchfall; interstitielle Nephritis mit starker Proteinurie, Oligurie, Anstieg der Retentionswerte, bis 10% ANV, Thrombopenie, Petechien 2. Puumala: Nephropathia epidemica mit günstiger Prognose (Deutschland >90%) 3. Sin Nombre, Bayoi, Black, Creek-Canal, New York, Andes: Hantavirus-kardiopulmonlaes Syndrom (HCPS): USA, Kanada, Südamerika; Fieber, Myalgien, Übelkeit, Abdominalbeschwerden, trockener Reizhusten, interstitielles Lungenödem, hämorrhagische Pneumonie, ARDS; im Labor atypische Lymphozyten Th: ggf. Versuch mit Ribavirin Letalität des HFRS 5-10%, des HPS ca. 50% |
|
|
Reiter Syndrom
|
Reaktive Arthritis
Zweiterkrankung nach gastrointetinalem Infekt (Yersinien, Salmonellen, Shigellen, Campylobacter) oder urogenitalem Infekt (Gonorrhö, Chlamydia trachomatis, Mykoplasmen) 60-80% der Patienten sind HLA-B27 positiv. Latenzzeit von 2-6 Wochen nach enteritischem oder urethritischem Infekt 1. Arthritis (oft asymmetrische, evtl. wandernde Oligoarthritis, bevorzugt der unteren Extremitäten 2. Urethritis 3. Konjunktivitis/ Iritis 4. Reiter Dermatose (Balanitis circinata, Aphten im Mundraum, Keratoma blenorrhagicum = schwielenartige, teils pustulöse Veränderungen an Hand- und Fußsohlen, psoriasiforme Hauveränderungen am ganzen Körper Th.: -Infektsanierung, wenn Erreger nachweisbar -NSAR, evtl. Kortikoide 80% der Fälle heilen innerhalb von 12 Monaten spontan aus. |
|
|
Q-Fieber
|
Weltweite Zoonose, Erreger Coxiella burnetii wird durch Scahfzecken von Tier zu Tier übertragen, Tiere erkranken nicht, Menschen stecken sich durch aerogene Staubinfektion an.
Coxiella burnetii = obligat intrazellulär, 2 Formen: SCV (Small cell variant; lange Überlebensfähigkeit in der Umwelt) und LCV (large cell variant; im Wirt) 50% der Fälle asymptomatisch -Plötzliches hohes Fieber -Retrobulbäre Kopfschmerzen -Atypische Pneumonie Ko.: -Neurologische Symptome -Granulomatöse Hepatitis -Myokarditis, Perikarditis, chronisch verlaufende Endokarditis bei persistierender Infektion -Chronic Fatigue Syndrome Th.: Doxycyclin Bei Endokarditis Doxy + Chinolon oder Rifa + Cipro über mehrere Jahre! |
|
|
Anthrax
|
Bacillus anthracis, Toxin- und Sporenbildner, Sporen sehr widerstandsfähig
Zoonose, beim Menschen selten, Missbrauch als Kampfstoff -Lungenmilzbrand (schwere Bronchopneumonie mit hohem Fieber) -Hautmilzbrand -Darmmilzbrand Sofortiger Therapiebeginn mit Ciprofloxacin oder Doxycyclin |
|
|
Adenoviren
|
-Keratokonjunktivitis epidemica
-Pharyngitis -Gastroenteritis -Pneumonie |
|
|
SARS
|
Schweres akutes respiratorisches Syndrom
Coronavirus, aerogene Tröpfcheninfektion Verdachtsfall: -Hohes Fieber und Husten oder Atembeschwerden und -enger Kontakt mit verdächtigem SARS-Fall oder -Aufenthalt in einem SARS-Befallsgebiet -Anstieg LDH, GOT Th.: Isolierstation, symptomatische Therapie, antivirale Substanzen Letalität altersabhängig Epidemie in China im Jahr 2003 |
|
|
Opportunistische Pneumonien
|
Viren: CMV, Herpes simplex, Varizella zoster
Pneumocystis-Pneumonie Systemische Pilzinfektionen: Candidiasis, Aspergillos |
|
|
Pneumocystis Pneumonie
|
Pneumocystis jiroveci, Schlauchpilz, persistiert bei der Mehrzahl der Menschen latent in der Lunge
Häufigste Erstmanifestation von AIDS, häufigste opportunistische Infektion bei AIDS Kl.: Dyspnoe, Tachypnoe, trockener Husten, Fieber Auskultation und Rö-Thorax initial meist unauffällig. Rö Tho.: Verminderung der Inspirationstiefe, basal betontes retikulonoduläres Muster mit Aussparung der Peripherie, alveoläre Verdichtungen mit Milchglasmuster, Progress bis zur weißen Lunge Frühdiagnose im CT, typisch: Subsegmentale Aussparungen Verlauf langsam oder perakut Th.: Cotrimoxazol für 21 Tage in hoher Dosierung; Reserve: Atovaquon, Pentamidin Primärprophylaxe bei AIDS bei T4 <200/yl, Sekundärprophylaxe bei überstandener Infektion (Cotrim oder Pentamidininhal.) |
|
|
Aspiration von Magensaft
|
Mendelson-Syndrom
Einsetzen der Symptomatik nach einer Latenzzeit von 2-12 Stunden: Bronchospasmus, bronchiale Hypersekretion, Dyspnoe, resp. Insuff. Ko.: Aspirationspneumonie durch Anaerobier, gramnegative Bakterien Th.: Clindamycin + Cephalosporin |
|
|
Systemische Pilzinfektionen
|
Europa: Candidiasis, Aspergillose, Kryptokokkose
Außerhalb Europa: Histoplasmose, Kokzidiomykose, Blastomykose Fakultativ pathogen: Candida, Aspergillus Obligat pathogen: Kryptokokkose, Blastomykose, Histoplasmose, Kokzidiomykose |
|
|
Antimykotika
|
Ampho-B: Mittel der Wahl bei lebensbedrohlichen Pilzinfektionen
Cave: Nierenwerte, Leberwerte, Panzytopenie Azole: Fluconazol (Diflucan)->Candida, Voriconazol (Vfend) -> Aspergillus Echinocandine: Caspofungin (Cancidas) -> Candida und Aspergillus |
|
|
Aspergillose
|
1. Allergische bronchopulmonale Aspergillose
2. Invasive pulmonale Aspergillose und Aspergillus-Pneumonie 3. Aspergillom |
|
|
Krankheitsphasen der Tbc
|
1. Primärtuberkulose (Erstinfektion)
a) Latente Tbc (erfolgreiche Eindämmung der Erreger; pos. Hauttest und post. Interferon-Gamma-Test aber Röntgen negativ) b) Progressive Primärtuberkulose 2. Postprimärtuberkulose (Reaktivierung) 3. Narbenstadium |
|
|
Primärturberkulose
|
a) latent (Konversion des Hauttest oder des Interferon-Gamma-Tests, Rö negativ)
b) manifest: -Primärkomplex (intrapulmonaler spezifischer Herd = Ghon-Herd, mit lokaler LK-Reaktion oder Infiltrate; Klinisch häufig asymptomatisch -Primärkaverne -Minimal Lesions (hämatogene Streuung in verschiedene Organe; Lunge: Simon´sche Spitzenherde) fakultativ: B-Symptomatik, Erythema nodosum Ko.: -Hiluslymphknoten-Tbc (-> ML-Syndrom) -Pleuritis -Meningitis -Miliar-Tbc -Käsige Pneumonie -Landouzy Sepsis |
|
|
Postprimärtuberkulose
|
Endogene Reaktivierung alter Organherde (z.B. Simon´ Spitzenherd -> Assmann´Frühinfiltrat, meist infra- oder retroklavikulär -> ggf. Einschmelzung zur Kaverne) oder (seltener) exogene Reinfektion
80% pulmonal 20% extrapulmonal (LK, Pleura, Urogenitaltrakt, Knochen, Verdauungstrakt, ZNS) Pulmonale Manifestationen: -Durch Reaktivierung eines alten Spitzenherdes: Assmann´Frühinfiltrat (infraklavikulär), -wenn keine Abheilung: Kaverne mit Bronchusanschluss (offene Tbc); Hämoptysen, Bronchustbc, käsige Pneumonie, Miliar-Tbc, Sepsis, Penumothorax, Karzinom, Aspergillom |
|
|
Pathogenese der Tbc
|
Mycobacterium tuberculosis, säurefeste Stäbchen.
Intrazelluläre Persistenz in mononukleären Phagozyten. Produktive Form: Immunmechanismen führen bei guter Abwehrlage zur Granulombildung: Innen Epitheloidzellsaum mit Langerhans´Riesenzellen zur Begrenzung der zentralen Nekrose, außen Lymphozytensaum. Exsudative Form: Exsudation und Nekrose (Verkäsung). |
|
|
MDR-Tbc
|
Multi-Drug-resistant Tbc:
Resistenz mindestens gegen INH und RMP |
|
|
Diagnose der Tbc
|
-Röntgen
-Bakteriologisch: Sputum, Magennüchternsaft, Bronchiallavage (PCR, Mikroskopie- Ziehl-Neelsen-Färbung, Kultur) -Tuberkulin-Hauttest: Intrakutantest nach Mendel-Mantoux: Induration von 5mm pos. bei pos. Rö-Bild und klin. Verdacht. Ab 15mm pos. bei Pat. ohne Risikofaktoren und negativem Rö-Bild. -Interferon-Gamma-Test, höhere Sensitivität und Spezifität als Hauttest Hauttest pos. 8 Wochen nach Infektion. |
|
|
Standardtherapie der unkomplizierten Tbc
|
Isoniazid 6 Monate
Rifampicin 6 Monate Pyrazinamid 2 Monate Ethambutol 2 Monate Möglich auch Streptomycin Kontrollen: Leber (INH, RMP, PZA) Augen (EMB) Nieren (SM, PZA) Ohren (SM) Polyneuropathie INH Komplizierte Tbc (AIDS, Immunsuppression, Rezidiv, Meningitis): 9-12 Monate |
|
|
Lues/ Syphilis
|
Treponema pallidum = spiralförmige Spirochäte, nicht anzüchtbar
A) Konnatale Lues: Hutchinson Trias: Tonnenzähne, Innenohrschwerhörigkeit, Keratitis parenchymatosa B) Erworbene Lues 1. Primärkomplex: Schmerzloses Ulcus durum (gerötet, nässend, hochinfektös, am Genitale) + geschwollene LK 2. Sekundärstadium: Hämatogene und lymphogene Aussaat 2-3 Monate nach Infektion: Roseolen, papulöse Syphilide, Condyloma lata, Handfächen, Fußsohlen, Plaques muqueses der Mundschleimhaut, Angina specifica; Iritis, Hepatitis, LK-Schwellungen 30% spontane Ausheilung 3. Tertiärstadium: 5-10 Jahre nach Infektion: Gummen in Knochen, Muskeln, Herz, Lunge, Magen, Darm; Haut; typisch: Mesaortitis syphilitica; Neurosyphilis: Tabes dorsalis, Argyll-Robertson-Phänomen, progressive Paralyse Di.: Dunkelfeldmikroskopie, Fluoreszenzmikrsokopie, TPHA (Treponema pallidum Hämagglutinationstest, Screeningtest), FTA-Abs-Test (Fluoreszenz-Treponema-Antikörper-Absorptionstest; Bestätigungstest) Th.: Penicillin, bei Allergie Doxycyclin oder Erythromycin |
|
|
Gonorrhoe
|
Neisseria gonorrhoeae, gramnegative Diplokokken
Frauen: Urethritis, Zervicitis, Ko.: PID, Fitz Hugh Curtis Syndrom Männer: Urethritis, Ko.: Prostatitis, Epidymitis Disseminierte Gonokokkeninfektion: Sepsis, Endokarditis, Meningitis Neugeborene: Eitrige Konjunktivitis Reaktive Arthritis (oft Monoarthritis des Kniegelenks) Einmaltherapie mit Cephalosporin II/ III |
|
|
Morbus Paget
|
Ostitis deformans Paget
Knochenerkrankung unklarer Genese mit erhöhten Knochenumbauvorgängen mit dem Risiko von Verformungen; nach Osteoporose die zweithäufigste Knochenerkrankung! Frühphase: Unkontrollierte Stimulation des osteoklastären Knochenabbaus Spätphase: Überschießender ingeordneter Knochenanbau -> aufgetriebener, mechanisch instabiler Knochen mit Neigung zu Frakturen -30% asymptomatisch -lokale Knochenschmerzen, evtl. erhöhte Temperatur über dem Knochen -Säbelscheiden Tibia -Zunahme des Kopfumfangs (Hut passt nicht mehr) -Arthrosen -Wurzelkompressionssyndrome -Schwerhörigkeit (Schallleitung oder Innenohr) -Nierensteine -Volumenbelastung des Herzens Th.: Bisphosphonate, Calcitonin (weniger effektiv), Calcium, Vit D |
|
|
"Hut passt nicht mehr"
|
Morbus Paget
Akromegalie |
|
|
Spannungspneu
|
Durch einen Ventilmechanismus gelangt bei jeder Inspiration Luft in den Pleuraraum, die bei Exspiration nicht entweichen kann
-> Druckanstieg im Pleuraraum -> Verlagerung des Mediastinums zur gesunden Seite mit Kompression der gesunden Lunge und Behinderung des venösen Rückstroms ->ZVD Anstieg -> Abfall des HZV |
|
|
Häufigste Gründe für Pleuraerguss
|
Transsudat:
1. Linksherzinsuffizienz 2. Lungenembolie 3. Leberzirrhose Exsudat: 1. Pneumonie 2. Malignom 3. Lungenembolie weitere: -Tuberkulose -subphrenischer Abszess (Pankreatitis) -SLE -Dressler-Syndrom (Postmyokardinfarkt) -Nephrotisches Syndrom, Urämie -Exsudative Enteropathie -Meigs-Syndrom |
|
|
Ursachen einer sekundären Kälteagglutinin-Krankheit
|
-NHL
-Infekte: Mykoplasmen, EBV, Listerien |
|
|
Marchiafava-Anämie
|
Erworbene klonale Erkrankung der pluripotenten hämatopoietischen Stammzelle mit Störung des Phophatidyl-Inositol-Glykan-Ankers (PIG-Anker):
-> Erythrozytenmembran unzureichend vor Komplement geschützt, bei Stress, Infekten oder morgens nach Abfall des Blut-pH Hämolyse ->Freies Hämoglobin bindet NO -> Kontraktionen der glatten Muskulatur und Thrombozytenaktivierung! -> Thrombembolie Trias: 1. Hämolyse 2. Thrombose 3. Zytopenie -Colafarbener Morgenurin, Gefahr Nierenversagen bei schweren hämolytischen Krisen -Thrombosen häufig an ungewöhnlicher Lokalisation (z.B. Pfortader, Lebervenen- Budd-Chiari-Syndrom, Milzvenen, Sinusvenen); auch Myokardinfarkt möglich -durch Vasokonstriktion bedingte Symptome: Kopfschmerzen, Dysphagie, abdominale Schmerzen, pulmonale Hypertonie, erektile Dysfunktion Di.: Zuckerwassertest, Durchflusszytometrie Ko.: Übergang in MDS oder AML, Chronische Niereninsuffienz, pulmonale HTN |
|
|
Therapie Marchiafava-Anämie
|
Kurativ: Allogene Stammzelltransplantation
Eculizumab (Anti-C5-mAb)-> Blockierung der terminalen Endstrecke der Komplementaktivierung durch Antikörper gegen C5 Cave: Erhöhtes Risiko für Meningokokken-Infektionen (-> Impfung!) Symptomatisch: -Transfusionen -Folsäure, B12, Eisen -bei hämolytischer Krise: Hydrierung, ggf. Infekttherapie, Kortikoide können versucht werden, Eculizumab -Antikoagulation -Hämatopoietische Wachstumsfaktoren |
|
|
Häufigste Todesursache bei Marchiafava-Anämie
|
Thrombosen
Weitere Komplikationen: -Übergang in MDS oder AML -Hämolytische Krise |
|
|
Klinik der akuten hämolytischen Krise
|
-Fieber, Schüttelfrost
-Ikterus, Hyperblilirubinämie -abdominelle Schmerzen, Rückenschmerzen, Kopfschmerzen -Hämoglobinurie mit bierbraunem Urin |
|
|
Hämochromatose/ Hämosiderose
|
Hämochromatose: Eisenablagerung mit Gewebeschädigung
Hämosiderose: Eisenablagerung ohne Gewebeschädigung |
|
|
Formen der Hämochromatose
|
Primäre Hämochromatose:
Mutation im HFE-Gen, verschiedene Mutationen mit unterschiedlicher Penetranz Sekundäre Hämochromatose: -hämolytische Anämie (z.B. Thalassämie), MDS -> erhöhte Eisenaufnahme im Darm, Transfusionen -alkoholische Siderose (vermehrte Eisenaufnahme) |
|
|
Klinik und Diagnose der Hämochromatose
|
-Leberzirrhose, HCC
-Diabetes mellitus ("Bronzediabetes") -dunkle Hautpigmentierung -Kardiomyopathie -hypophysärer Hypogonadismus bei juveniler Hämochromatose -schmerzhafte Arthropathie (typisch: Fingergrundgelenke) Di.: -Ferritin hoch, Transferrinsättigung hoch -Leberbiopsie (Berliner-Blau-Färbung) -HFE-Gendiagnostik |
|
|
Therapie der Hämochromatose
|
-Eisenarme Diät
-Aderlasstherapie -Eisenchelatoren (Deferoxamin) bei KI gegen Aderlässe, bei transfusionsbedingter Hämochromatose |
|
|
DD Megaloblastäre Anämie
|
Mangel an Vit. B12 und/oder Folsäure mit DNS-Synthese-Störung und Kernreifungsstörung der Myelopoese
Vit. B12-Mangel -mangelnde Zufuhr bei streng vegetarischer Kost -Mangel an Intrinsic Factor (Z.n. Magenresektion, perniziöse Anämie) -Malabsorption bei intestinalen Erkrankungen -Fischbandwurm -bakterielle Überwucherung ("blind-loop-Syndrom) Folsäure-Mangel: -Mangelernährung (Alkoholiker) -erhöhter Bedarf (Schwangerschaft, Hämolyse) -Malabsorption -Medikamente: FS-Antagonisten, Azathioprin, Triamteren, Phenytoin |
|
|
Bei welchen hämatologischen Erkankungen ist die LDH erhöht?
|
-Hämolyse
-megaloblastäre Anämie |
|
|
Formen der respiratorischen Insuffizienz
|
1. Hypoxämisches Versagen = respiratorische Partialinsuffizienz (Lungenparenchymversagen)
2. Hyperkapnisches Versagen = respiratorische Globalinsuffizienz (Atempumpversagen) |
|
|
Bronchiektasen
|
Sackförmige oder zylindrische Ausweitungen der mittleren und distalen Bronchien, meist basal in den UL
angeboren (z.B. zystische Fibrose) oder erworben (COPD, Bronchusstenosen, Tbc) Mukostase und rezidivierende bakterielle Infekte führen zu produktivem Husten: Sputum oft dreischichtig (Schaum, Schleim, Eiter), maulvolle Expektorationen Ko.: -Lungenblutung -rezidivierende bronchopulmonale Infekte -Abszess -bakteriell metastatische Herde -Pilzbesiedlung -Amyloidose -resp. Insuffizienz |
|
|
Lungenblutung
|
Hämoptoe: massives Aushusten von Blut
Hämoptyse: leichte Blutbeimischung im Auswurf -Tbc -Bronchialkarzinom -Bronchiektasen -Lungeninfarkt -Trauma -Bronchitis, Pneumonie -Good-Pasture-Syndrom -Wegener Granulomatose |
|
|
Atelektasen
|
luftleeres Lungengewebe ohne entzündliche Veränderungen
1. Obstruktionsatelektasen (Tumor, Schleimpfropf, Fremdkörper) 2. Kompressionsatelektasen (z.B. basale Plattenatelektasen bei Pleuraerguss) 3. Entspannungsatelektasen (Pneumothorax) |
|
|
COPD Stadienteinteilung
|
I leicht FEV1 >= 80 v.S.
II mittel FEV1 50-79% v.S. III schwer FEV1 30-49% v.S. IV sehr schwer FEV1 <30% v.S. oder <50% und pO2 <60mmHg, pCO2>50mmHg Beim Emphysem ist der FEV1-Wert nicht geeignet zur Stadieneinteilung. |
|
|
Def. chronische Bronchitis
|
In 2 aufeinander folgenden Jahren während mindestens 3 aufeinander folgener Monate Husten und Auswurf (produktiver Husten)
|
|
|
Häufige Keime bei exazerbierter COPD
|
-Hämophilus influenzae
-Pneumokokken -Moxarella catarrhalis Bei fortgeschrittenen Fällen Wandel des Erregerspektrums: Enterobakterien, Proteus, Klebsiellen, Pseudomonas) |
|
|
Wegenersche Granulomatose
|
Nekrotisierende Vaskulitis vorwiegend der kleinen bis mittelgroßen Gefäße mit ulzerierenden nicht verkäsenden Granulomen
Kl: Lokal begrenztes Initialstadium: -Chronische Rhinitis, Sinusitis, Otitis; blutig borkiger Schnupfen; evtl. Sattelnase, Septumperforation -Ulzerationen im Oropharynx -Lungenrundherde, evtl. mit Einschmelzungen, evtl. subglottische Larynxstenose Vaskulitische Generalisation: Pulmo-renales Snydrom: -alveoläre Hämorrhagie -(rapid progressive) Glomerulonephritis evtl. Episkleritis, Arthralgien, Myalgien, ZNS-Symptome, Mononeuritis multiplex, Fieber, Nachtschweiß Gewichtsverlust Lab.: -hohe BSG, cANCA |
|
|
Therapie der Wegener Granulomatose und des Churg-Strauss-Syndrom
|
M. Wegener Initialstadium: Therapieversuch mit Cotrimoxazol
Beide: -Remissionsinduktion mit Prednisolon + Cyclophosphamid -Erhaltungstherapie mit MTX oder Azathioprin, Reduktion der Prednisolondosis |
|
|
ACR-Kriterien M. Wegener
|
1. Enzündung in Nase oder Mund mit Ulzera und purulenter nasaler Sekretion
2. Knoten, Infiltrationen oder Kavernennachweis im Rö-Thorax 3. Pathologisches Urinsediment mit Mikrohämaturie oder Erythrozytenzylindern 4. Bioptisch nachgewiesene granulomatöse Entzündung in art. Gefäßwänden 2/4 |
|
|
ANCA-assoziierte Vaskulitiden der kleinen Gefäße
|
1. Wegener Granulomatose
2. Churg-Strauss-Vaskulitis 3. Mikroskopische Panarteriitis |
|
|
nicht-ANCA-assoziierte Vaskulitiden der kleinen Gefäße
|
1. Purpura Schoelein Hennoch
2. Vaskulitis bei essentieller Kryoglobulinämie 3. Kutane leukozytoklastische Angiitis |
|
|
Vaskulitis mittelgroßer Gefäße
|
1. Klassische Panarteriitis
2. M. Kawasaki |
|
|
Vaskulitis großer Gefäße
|
1. Riesenzell Arteriitis (A. temporalis)
2. Takayasu-Arteriitis |
|
|
Mikroskopische Polyangiitis
|
Die MPA ähnelt klinisch der Wegenerschen Granulomatose, hsitologisch fehlt jedoch die granulomatöse Entzündung.
Kl.: -Nierenbeteiligung -> bestimmt die Prognose -pulmonale Vaskulitis, evtl. mit diffuser pulmonaler Hämorrhagie -Hautveränderungen: subkutane Knötchen, palpable Purpura Lab.: cANCA |
|
|
NET des Ileums und der Appendix
|
Epitheliale TU der enterochromaffinen Zellen des DNES (diffuses neuroendokrines System) mit Produktion von
1. Serotonin -> Diarrhö und Endokardfibrose (Hedinger-Syndrom, TK und PK) 2. Kallikrein -> Umwandlung von Kininogen zu Bradykinin -> Flush 3. Tachykinine 4. Prostaglandine -> Asthmaanfälle Lokalisation: 90% intestinal (50% Appendix, 15% distales Ileum) 10% Bronchuskarzinoide Mit Ausnahme des Bronchuskarzinoids Karzinoidsyndrom erst durch Lebermetastasen (Serotonin wird durch Monaminoxidasen der Leber abgebaut) weitere Symptome: Teleangiektasien, pellagraartige Hautveränderungen, Cushing-Syndrom durch ektope ACTH-Bildung Lab.: 5-Hydroxyindolessigsäure im 24-h-Urin (Abbaupr. von Serotonin), Serotonin, Chromogranin A im Serum Th.: Operation, Octreotid (Somatostatinanalogon, hemmt Hormonprod.), Alpha-Interferon; symptomatisch: Methysergid (Serotoninantagonist) |
|
|
Bei welchen Erkrankungen kommen Asthmaanfälle sekundär vor?
|
1. Karzinoid-Snydrom
2. Churg-Strauss-Snydrom 3. Sarkoidose |
|
|
Abgrenzung Asthma bronchiale - COPD
|
Typisch für Asthma bronchiale sind:
1. Hyperreagibilität des Bronchialsystems 2. Reversibilität: Besserung von FEV1 > 20% nach Gabe kurzwirksamer Beta-Mimetika oder Normalisierung der Lungenfuntkion. Gelegentlich Sicherung der Diagnose nur mittels oraler Behandlung mit 20-30mg Prednisolon für 7-10 Tage: Normalisierung der Lufu = Asthma bronchiale. Besserung FEV1 >25% meist Kombination aus COPD und Asthma bronchiale |
|
|
Welche Formen des Asthma bronchiale unterscheidet man?
|
1. Extrinsic Asthma:
-allergisch -Beginn meist in der Kindheit, oft zunächst als allergische Rhinitis -erhöhtes IgE, Eosinophilie 2. Intrinsic Asthma: -Asthma durch respiratorische Infekte -gelegentlich Analgetika-Intoleranz -Beginn meist im Erwachsenenalter -mögl. Ursache auch GERD -> bei Verdacht ex juvantibus Therapie mit PPI für 6 Monate |
|
|
Asthma - Pathophysiologie
|
-Entzündungsreaktion der Bronchialschleimhaut, ausgelöst durch Allergene oder Infekte: Mastzellen, Eosinophile, T-Lymphozyten.
IgE-vermittelte Sofortreaktion ->Degranulation von Mastzellen mit Freisetzung von Histamin, Leukotrienen, Bradykinin -> endobronchiale Obstruktion; daneben auch IgG-vermittelte Spätreaktion (nach 6-12 Stunden) möglich. -Bronchiale Hyperreagibilität -endobronchiale Obstruktion durch Bronchospasmus, Schleimhautödem, Hypersekrektion eines zähen Schleims, Umbauvorgänge der Bronchien |
|
|
Welche Medikamente sind beim Asthmapatienten mit Vorsicht einzusetzen?
|
-Beta-Blocker
-ACE-Hemmer -ASS/ NSAR -Parasympathomimetika (Pilocarpin, Carbachol) |
|
|
Vocal Cord Dysfunction
|
Spasmen der Stimmlippen, die mit Stridor und oft perakuter Atemnot einhergehen können. Dyspnoe kurz andauernd (1-2 Minuten)
Aufklärung über Harmlosigkeit |
|
|
Schweregradeinteilung des Asthma bronchiale
|
GINA 2006
TS:Tagesymptome NS: Nachsymptome E: Exazerbationen 1. Intermittierendes A.: TS. < 1x/W, NS <= 2x/M, E kurz, FEV1>=80% 2. Mild persitierendes A.: TS >1/W, <1x/T, NS >2x/M, E Beeinträcht. von Schlaf und Aktivität, FEV1 >=80% 3. Mittelschwer persistierendes A.: Täglich Symptome, NS >1/W, E Beeinträcht. von Schlaf und Aktivität, FEV1 60-80% 4. Schweres persistierendes A.: Tägliche Symptome, häufige Nachtsymptome, körperliche Aktivität vermindert, FEV1 < 60% |
|
|
Welche Leitlinien sind für die Asthmatherapie maßgeblich?
|
GINA
Global Initiative for asthma |
|
|
Kann Asthma bronchiale zum Cor pulmonale oder Emphysem führen?
|
Nein!
|
|
|
Therapie des Asthma bronchiale
|
Stufenschema GINA
St. 1: RABA St. 2: ICS St. 3: ICS + LABA St. 4: ICS hochdosiert + LABA, ggf. + Theophyllin/ LTRA St. 5: zusätzlich orale Steroide, ggf. Omalizumab Beurteilung ob A. danach kontrolliert, teilweise kontrolliert oder nicht kontrolliert Köhler: Grundlage = Inhalative Steroide, zu Beginn orale + inhalative Steroide, zusätzlich nasale Steroide RABA: Rapid Acting Beta-2 Agonist LABA: Longe Acting Beta-2 Agonist ICS: Inhalatives Kortikoid |
|
|
Therapie des exazerbierten Asthma bronchiale
|
Prednisolon 20-30mg - 0- 10-20mg, 1 Woche, dann 1 Woche reduziert, dann absetzen
Akuter Asthmaanfall: 50mg Prednisolon p.o. (i.v. wirkt nicht schneller) |
|
|
Ab welcher Therapiedauer ist ein Ausschleichen von Steroiden notwendig?
|
2-4 Wochen
|
|
|
Wie lange brauchen inhalative Steroide, bis sie ihre volle Wirksamkeit entfalten, wie lange wirken sie nach dem Absetzen noch nach?
|
Jeweils 4 Wochen
|
|
|
COPD Pathophysiologie
|
-Chronische Entzündung mit CD8-Lymphos, Neutrophilen, Makrophagen im Bereich der kleinen Atemwege, die durch inhalative Noxen ausgelöst wird.
-Ungleichgewicht zwischen Proteasen und Proteaseinhibitoren mit Destruktion des Lungengewebes -Obstruktion durch: Remodeling (Fibrosierung), Parenchymverlust und bronchiale Instabilität, mukoziliäre Dysfunktion, Hypersekretion Typisch: Im Verlauf zunehmende Fixierung der Obstruktion, Bronchialkollaps bei forcierter Exspiration -> Überblähung |
|
|
Unterschiedliche Phänotypen der chronischen Bronchitis
|
1. COPD -> Blue Bloater:
-Überblähung -Überlastung der Atemmuskulatur -hyperkapnisches Versagen 2. Emphysem -> Pink Puffer -Reduktion der Gasaustauschfläche -Hypoxämisches Versagen |
|
|
Alpha-1-Antitrypsin-Mangel
|
Physiologisches Gleichgewicht zwischen Proteasen (z.B. Elastase) aus neutrophilen Granulozyten und Antiproteasen (Alpha-1-Antitrypsin) gestört durch Mangel AAT
1. Homozygote Form: -AAT < 50mg/dl -Hepatitis, Leberzirrhose ab Kindheit -Fast alle entwickeln im Erwachsenenalter ein Lungenemphysem 2. Heterozygote Form -7% der Bevölkerung -AAT 50-250mg/dl -Entscheidend für den Beginn des Lungenemphysems sind auslösende pulmonale Noxen (Infekte, Rauchen, Staub) Zigarettenrauch inaktiviert AAT! |
|
|
Begleiterkrankungen COPD
|
-Osteoporose
-Muskelatrophie -Depression -Cor pulmonale -pulm. Hypertonie -KHK |
|
|
Therapie der COPD
|
I Leichtgradig: RABA/ Anticholinergika bedarfsweise
II Mittelgradig: Dauertherapie mit LABA/ Anticholinergika III Schwergradig: Zusätzlich Therapieversuch mit inhalativen Steroiden, evtl. Theophyllin IV Sehr schwer: Zusätzlich O2-Langzeit-Therapie bei resp. Insuffizienz Köhler: -Steroide nur dann, wenn zusätzlich Zeichen eines Asthma bronchiale. -Pat. mit Lungenüberblähung reagieren oft besser auf Anticholinergica als Pat. mit reiner Obstruktion. -Orale Steroide nur bei Exazerbation (30-50mg/d!) -Theophyllin und LABA nur morgens geben, reicht -Theophyllin soll mukoziliäre Clearance verbessern -Inhalation mit Kochsalz -Ggf. Inhalation mit Gentamicin |
|
|
Ernährung hyperkapnischer Patienten
|
Mehr Fette als Kohlenhydrate -> geringere CO2-Produktion
|
|
|
Erkennung der Exazerbation der COPD
|
Anthison-Kriterien:
1. Zunahme des Hustens und des Sputums 2. Zunahme der Luftnot 3. Verfärbung des Sputums in Richtung grün/gelb |
|
|
Therapie der Exazerbation einer COPD
|
-orale Steroide (30-50mg/d, 2 Drittel morgens, 1 Drittel nachmittags, für 14 Tage)
-Antibiotikatherapie: Amoxicillin + Clavulansäure oder Levofloxacin, Moxiflocacin evtl. reichen auch Tetrazykline, Cotrim oder Makrolide In der Klinik: z.B. Clinda + Cipro oder Fortum/Tazobac +Clinda (Köhler) -Kein Theophyllin i.v., keine Beta-Mimetika i.v. |
|
|
Ursachen Lungenfibrose
|
1. Infektionen (z.B. Pneumocystis)
2. Inhalative Noxen: Pneumokoniosen, exogen allergische Alveolitis 3. nichtinhalative Noxen (Bleomycin, Amiodaron, Herbizide) 4. Ionisierende Strahlen (Strahlenpneumonitis) 5. Chron. Stauungslunge, ARDS, Fluid Lung bei Niereninsuffzienz 6. Systemerkrankungen (Sarkoidose, RA, Kollagenosen, Vaskulitiden, Speichkrankheiten) 50% idiopathische Lungenfibrose |
|
|
Pulmonale Histiozytose X
|
Granulomatöse Entzündung des Lungeninterstitiums, Granulome aus Histiozyten, eosinophilen Leukozyten, Lymphozyten, Plasmazellen, Langerhans´ Zellen
Bei Kindern häufig disseminiert, bei Erwachsenen (oft Raucher) pulmonale Manifestation: Interstitielle Lungenfibrose und Ausbildung von Lungenzysten (Gefahr Pneumothorax durch Zystenruptur) |
|
|
Silikose
|
Quarzstaublungenerkrankung; Metallhütten, Walzwerke, Steinbruch, Glas-/Porzellan-/Keramikindustrie, Sandstrahlarbeiten
Quarzpartikel werden von Makrophagen aufgenommen -> Makrophagenzerfall -> fibroblastische Reizwirkung -> Knötchen Kl.: 1. Infektanfälligkeit, Siliko-Tbc 2. COPD, Lungenemphysem, Cor pulmonale 3. Lungenkrebsrisiko 4. Erhöhtes Risiko für progressive systemische Sklerose "Eierschalenhilus" |
|
|
Asbestose
|
Asbest=Faserförmige kristallisierte silikatische Mineralien. 95% Chrysotil (Weißasbest). Kritische Abmessung karzinogener Faser: Länge >5ym, Durchmesser <3ym
Asbestfasern driften in Richtung Pleura und akkumulieren subpleural. Kl.: 1. Asbestose (asbestinduzierte Lungenfibrose) 2. Pleuraplaques, Asbestpleuritis 3. Lungenkarzinom 4. Mesotheliom 5. Larynkarzinom |
|
|
Risikofaktoren für Bronchialkarzinome
|
1. Zigarettenrauchinhalation -> für 85 % aller BC verantwortlich. 40py -> 10-faches Risiko, bei Beginn des Rauchens in der Jugend bis 30-faches Risiko.
2. Berufliche Karzinogene (5% aller BC, davon 90% durch Asbest) 3. Umweltkarzinogene (Radon, Passivrauchen, Industrieabgase) 4. Narbenkarzinom, Kavernenkarzinom 5. Genetische Disposition |
|
|
Histologie der Bronchialkarzinome
|
1. SCLC: meist zentral lokalisiert, schnelles Wachstum, bei Diagnose meist schon metastasiert, "oat cell carcinoma", paraneoplastische Sekretion von Hormonen
2. NSCLC -Adenokarzinom: Oft peripher lokalisiert, häufig bei Nichtrauchern, seltene Sonderform: Alveolarzellkarzinom -Plattenepithelkarzinom: Vorwiegend zentral lokalisiert -Großzellige Lungenkarzinome (adenosquamöses Karzinom, sarkomatoides Karzinom, Karzinoid) |
|
|
Häufigste Fernmetastasen-Lokalisationen bei BC
|
Leber
Gehirn Nebennieren Skelett |
|
|
TNM-Klassifikation Bronchialkarzinom
|
TIS Carcinoma in situ
T1a <= 2cm T1b >2-3cm T2 Hauptbronchus >=2cm von Carina, Invasion von viszeraler Pleura, part. Atelektase T2a >3-5cm T2b >5-7cm T3 >7cm, Brustwand, Zwerchfell, Perikard, mediast. Pleura, Hauptbronchus <2cm von Carina, totale Atelektase, separate TU-Herd(e) im selben Lappen T4 Mediastinum, größere Gefäße, Carina, Trachea, Ösophagus, Wirbelkörper, separate(r) Herd(e) in einem ipsilateralen Lappen N0 N1 Ipsilaterale peribronchiale/hiläre LK N2 Ipsilaterale mediastinale/subkarinale LK N3 Kontralaterale mediastinale, hiläre, ipsi- oder kontralaterale Skalenus- oder supraklavikuläre LK M0 M1a Separate(r) Tumorherd(e) in einem kontralateralen Lappen, Pleurametastasen, maligner Pleura- oder Perikarderguss M1b Fernmetastasen |
|
|
UICC-Stadien Bronchialkarzinom
|
Stadium 0 TIS N0 M0
St. IA T1a, T1b N0 M0 St. IB T2a N0 M0 St. IIA T2b N0 M0; T1a t1b, T2a N1 M0 St. IIB T2b N1 M0, T3 N0 M0 St. IIIA T1a,b, T2a,b N2 M0, T3 N1,N2 M0, T4 N0,N1 M0 St. IIIB T4 N2 M0, jedes T, N3, M0 St. IV Jedes T Jedes N M1 |
|
|
Stadieneinteilung SCLC
|
Very limited disease (St. I): Auf die Lunge begrenzter Primärtumor ohne mediastinalen LK-Befall. T1- T2 N0-1
Limited disease (St. II-III): Befall eines Hemithorax mit oder ohne ipsi- oder kontralaterale mediastinale oder ipsilaterale supraklavikuläre LK-Metastasen mit/ohne ipsilateralen Pleuraerguss Extensive disease: (St. IV) |
|
|
Pancoast Tumor
|
Peripheres BC der Lungenspitze, das Pleurakuppe und Thoraxwand arrodiert und dabei Halssympathikus und zervikale Nervenwurzeln schädigt
-> Plexuxneuralgie (Armschmerzen), Interkostalneuralgie -> Horner Snydrom (Miosis, Ptosis, Enophthalmus) -> Armschwellung (Lymph- und Venenstau) |
|
|
Paraneoplastische Syndrome, insbesondere beim SCLC
|
1. Paraneoplastische Endokrinopathien:
-Cushing-Syndrom (ektope ACTH-Produktion) -SIADH -Tumorhyperkalzämie (PTHrP) -Hypoglykämie (Insulin-like-factor) 2. Paraneoplastische Neuropathien und Myopathien -Lambert-Eaton-Syndrom (myasthenieartige Schwäche) -Kleinhirndegeneration -Polymyositis, Dermatomyositis 3. Thrombozytose 4. Pierre-Marie--Bamberger-Syndrom (hypertrophe pulmonale Osteoarthropathie: Trommelschlegelfinger, Uhrglasnägel, Gelenkschmerzen) |
|
|
Karzinomverdächtige Faktoren eines Lungenrundherdes
|
-Raucheranamnese
-Alter >40 Jahre -Fehlende Verkalkung -Spiculae, die vom Rundherd ins Lungenparenchym ausstrahlen -Größenzunahme im Vergleich zu Voraufnahmen |
|
|
Operabilität bei BC
|
TLCOc muss nach OP>40% sein.
FEV1 muss nach OP>40% sein. Pro Lappen verliert ein Patient 20% seiner Kapazität, pro Lungenflügel 50%. |
|
|
Therapie SCLC
|
Limited disease (bis T2N0M0): Resektion mit kurativer Zielsetzung und Radiochemotherapie
PE (Cisplatin, Etoposid) Radiatio mit 40Gy, prophylaktische Schädelbestrahlung Extensive disease: Polychemotherapie (ACO - Adriamycin, Cyclophosphamid, Vincristin) oder CEV (Carboplatin, Etoposid, Vincristin) oder PE Radiatio nur bei Hirn- oder Skelettmetastasen sowie bei oberer Einflusstauung |
|
|
Therapie NSCLC
|
bis St. IIIA radikale Operation mit LK-Dissektion
evtl. adjuvante oder neoadjuvante Chemotherapie Ab St. IIIB Radiochemotherapie oder kombinierte Chemotherapie, evtl. Bevacizumab bei Adeno-Ca, bei pos. EGF-Rezeptormutation Gefitinib |
|
|
Stevens Johnson Syndrom
|
Immunologische, T-Zell-vermittelte Reaktion mit Apoptose der Keratinozyten, meist durch Medikamente induziert.
Akuter Beginn mit schwerem Krankheitsgefühl, hohen Temperaturen, Rhinitis, Erythemen und schmerzhaften Blasen der Schleimhäute, Konjunktivitis. Maximalvariante: Lyell-Syndrom (toxische epidermale Nekrolyse) Letalität bis 6%,bei Lyell-Syndrom bis 50% |
|
|
Osteoporose
|
Verminderte Knochenmasse und Verschlechterung der Mikroarchitektur
95% Primäre Osteoporose: -Typ I (postmenopausale Osteoporose, 50 - 70 J, spongiosabetont) Typ II (senile Osteoporose, > 70J, Spongiosa plus Kompakta) 5% Sekundäre Osteoporose: Endokrin (Hyperkortisolismus, Hypogonadismus, Hyperthyreose, pHPT), Malabsorption, Immobilisation, Medikamente (Kortikoide, PPI, Antiandrogene, Antiepileptika, Aromtasehemmer, Glitazone) Risikofaktoren: Nikotinkonsum, Immobilität, Untergewicht, Vit D, Ca-Mangel, späte Menarche, frühe Menopause Fast-loser-Pat.: >3%/J Verlust Knochenmasse, typisch für frühe postmenopausale OP Slow-loser-Pat.: <3%/J, typisch für späte postmenopausale OP |
|
|
Klinik und Diagnostik der Osteoporose
|
-Knochenschmerzen, bes. Rücken
-Spontanfrakturen -Gibbus, Körpergrößenabnahme -Tannenbaumphänomen Osteodensitrometrie: DXA (Dual-X-Ray-Absortiometrie -> Messung der Flächendichte des Knochenmineralgehalts (g/cm2) T-Score: Standardabweichung vom Mittelwert der peak bone mass gesunder Menschen im Alter von 30 J T-Scores in Abhängigkeit von Lebensalter und Geschlecht, die im Mittel mit einem 30%igen Frakturrisiko für WK- und prox. Femurfx in 10 J assoziiert sind: Tabelle Rö der WS: Zunächst treten an den WK-Deckplatten und vertikale Trabekel hervor, im Verlauf Fischwirbel, Keilwirbel DD: sekundäre Osteoporose (TSH, Testosteron, Östrogene) -Plasmozytom, Knochenmetastasen, pHPT, Osteomalazie |
|
|
Therapie der Osteoporose
|
Medikamentöse Therapie, wenn 10-Jahresrisiko für WK- und prox. Femurfrakturen >30%
-Bisphosphonate (z.B. Alendronsäure) -> Hemmung der Osteoklasten -Strontiumranelat (Protelos): hemmend auf Osteoklasten, stimulierend auf Osteoblasten; NW: GI-Symptome, Hautreaktionen bis zu Stevens-J.-S., Thrombembolie -Raloxifen (Evista): selektiver Östrogenrezeptormodulator (auch Risiko für Mamma-Ca) -Denosumab (Prolia): Humaner klonaler AK, Inhibition der Osteoklasten -PTH (Teriparatid - Forsteo, rekombinantes PTH - Preotact): s.c.-Anwendung führt zu Knochenaufbau -Östrogen |
|
|
Osteomalazie und Rachitis
|
Osteomalazie: Ungenügende Mineralisation des Osteoids nach Abschluss des Knochenwachstums
Rachitis: Bevorzugter Befall der metaphysären Wachstumszone beim Kind -Vit D- Mangel -Störung des Vit D-Stoffwechsels (auf Leberebene mit mangelhafter Bildung von 25-OH-D3 - Calcifediol) oder auf Nierenebene (mangelhafte Bildung von 1,25-(OH)2-D3 - Calcitriol) Labor: Bei Malabsorption Hypophosphatämie und Verminderung von 25(OH)-D3; bei Niereninsuffizienz Hyperphosphatämie und Verminderung von 1,25(OH)2-D3 Kl.: -Skelettschmerzen -Knochenverbiegungen (o-Beine) -Adynamie -Watschelgang (Schwächung der Glutealmuskulatur) -Rachitis Rosenkranz (umschriebene Schwellung der Rippen an der Knochen-Knorpel-Grenze) -Neigung zu Tetanie -Looser´Umbauzonen - bandförmige Aufhellungen quer zur Längsachse des Knochens) |
|
|
Welche laborchemischen Faktoren haben Einfluss auf das Gesamtkalzium?
|
-Albumin: Abfall des Albumins bewirkt eine Erniedrigung des Gesamt-Ca
-pH: Alkalose-> niedriges Ca Azidose -> erhöhtes Ca |
|
|
Vitamin D
|
7-Dehydrocholesterol (Haut) durch UVB -> Cholecalciferol (Vit D3) (Nahrung) ->25-OH-D3 (25-Hydroxycolecalciferol (Leber) ->1,25(OH)2-D3 (Calcitriol=Vit-D-Hormon)
Ein niedriger Phosphatspiegel fördert die Calcitriolbildung. Calcitriol fördert die enterale Resorption von Calcium und Phosphat. |
|
|
Regulation der von Calcium und Phosphat
|
PTH: Erhöhung von Calcium durch Stimulation der tubulären Ca-Reabsorption und Stimulation der Ca-Resorption aus dem Knochen; Verminderung des Phosphat-Spiegels durch Hemmung der tubulären P-Reabsorption; außerdem Stimulation der Vit-D Synthese in der Niere
Vit-D3: Erhöhung von Ca und P durch Förderung der Resorption von Ca und P aus dem Darm und Förderung der Knochenmineralisation Calcitonin: Verminderung von Ca durch Hemmung der Osteoklasten Blut pH: Alkolose -> Verminderung des Ca (Hyperventilationstetanie!) |
|
|
Tertiärer HPT
|
Im Verlauf eines sekundären HPT kann sich eine Hyperkalzämie entwickeln (keine Autonomie sondern Missverhältnis zwischen PTH-Sekretion und Bedarf, z.B. nach Nierentransplantation)
|
|
|
Hypoparathyroidismus
|
Unterfunktion der Epithelkörperchen durch
-Z.n. Schilddrüsenoperation -idiopathisch -Di-George-Syndrom: Aplasie von Nebenschilddrüsen und Thymus Kl.: 1.Hypokalzämische Tetanie, Parästhesien, Pfötchenstellung, Stimmritzenkrampf -Chvostek´Zeichen: Beim Beklpfen des N. fascialis im Bereich der Wange wird ein Zucken des Mundwinkels ausgelöst Trousseau´Zeichen: Blutdruckmanschette, einige Minuten art. Mitteldruck -> Pfötchenstellung 2. Haar- und Nagelwuchsstörungen 3. Reizbarkeit, depressive Verstimmung Di.: -Hypokalzämie -Hyperphosphatämie -normales Krea -normales Albumin -vermindertes PTH |
|
|
DD Hypokalzämie
|
-Akute Pankreatitis
-Malabsorption -Peritonitis -Heilphase einer Osteomalazie oder Rachitis -Niereninsuffizienz |
|
|
Pseudohypoparathyroidismus und Pseudo-Pseudo-Hypopharathyroidismus
|
Familiär gehäuft, verschiedene Störungen in der PTH-Signalkette (PTH ist erhöht).
Typische körperliche Stigmata: Verkürzung von Mittelhand- und Mittelfußknochen, gedrungener Körperbau. Pseudo-Pseuo-H. wenn körperliche Stigmata ohne Kalziumstörung |
|
|
Nebennierenrinde - Welche Hormone werden (wo) synthetisiert?
|
1. Zona glomerulosa: Mineralkortikoide (Aldosteron)
2. Zona fasciculata: Glukokortikoide (Kortisol) 3. Zona reticularis: Androgene (Dehydroepiandrosteron) Alle werden aus Cholesterin synthetisiert. |
|
|
RAAS
|
Renin-Angiotensin-Aldosteron-System
Na-Mangel, Hypovolämie, verminderte Nierendurchblutung -> Ausschüttung von Renin aus den juxtaglomerulären Zellen der Niere -> Umwandlung von Angiotensinogen (Leber) zu Angiotensin I. Durch ACE (Angiotensin-Converting-Enzyme der Lunge) -> Angiotensin II: 1. Vasokonstriktion, RR-Steigerung 2. Freisetzung von Aldosteron in der NNR -> Na-Retention und Wasserretention |
|
|
Conn-Syndrom
|
Primärer Hyperaldosteronismus
2/3 IHA idiopathische bilaterale Hyperplasie der Zona glomerulosa, meist normokaliämisch 1/3 APA Aldosteron-produzierende Adenome der NNR, häufig mit Hypokalämie Kl.: Meist nur art. Hypertonie (häufigste Ursache einer sekundären Hyptertonie) <1/3 Trias: 1. Hypertonie 2. Hypokaliämie 3. Metabolische Alkalose Labor: -Aldosteron hoch -Renin niedrig Di.: -Screeningtest: Aldosteron/Renin-Quotient (ARQ) erhöht -Bestätigung: Kochsalzbelastungstest fehlende Aldosteronsuppression (2l 0,9% NaCl über 4h, Plasmaaldosteron >10ng/dl) -Orthostasetest (Anstieg von Aldosteron >30% spricht für bilaterale Hyperplasie, Abfall für Adenom) |
|
|
DD Polyurie + Polydipsie
|
1. Diabetes mellitus
2. Hyperkalzämie 3. Hypokaliämie 4. Diabetes insipidus |
|
|
Hypoaldosteronismus
|
1. Primär: erhöhter Reninspiegel (z.B. M. Addison)
2. Sekundär: niedriger Reninspiegel (renal tubuläre Azidose Typ IV; z.B. bei Diabetes mellitus, medikamentös) Hyponatriämie Hyperkaliämie Azidose |
|
|
Ätiologie Lungenödem
|
1. Kardial: Linksherzinsuffizienz mit Druckanstieg im Lungenkreislauf, z.B. hypertensive Krise
2. Nichtkardiales Lungenödem: -Fluid Lung bei Niereninsuffizienz -Postexpansionsödem (nach Pleurapunktion) -Höhenlungenödem -Anapyhlaktischer Schock -Toxisch: Reizgase |
|
|
EKG-Zeichen der Rechtsherzbelastung
|
-SI QIII Typ, SI SII SIII Typ
-p dextroatriale -Sinustachykardie, Vorhofflattern, Vorhofflimmern -(in)kompletter RSB -T-Negativierung / ST-Senkungen in V1-V3 -Verschieben der Übergangszone nach links Rechtsherzhypertrophie: -SV5 + RV1 >1,05mV (Sokolow-Lyon-Index) |
|
|
Langzeitsauerstofftherapie bei pulm. HTN
|
Indikation: PO2 in Ruhe <=55mmHg oder PO2 in Ruhe <=60mmHg und Cor pulmonale oder Polyglobulie
Ziel: Sichere Anhebung des PO2 auf > 60mmHg unter Ausschluss eines bedrohlichen CO2-Anstiegs O2-Therapie mindestens 16 Stunden pro Tag ->Pulmonalisdrucksenkung und Verbesserung der Überlebenszeit |
|
|
Spezifische Therapie bei pulmonaler Hypertonie
|
1. Langzeit-Sauerstofftherapie
2. Medikamente: -Kalziumantagonisten (Wirksamkeitsnachweis im Rechtsherzkatheter) -Prostanoide: Treprostinil s.c. oder Illoprost inhalativ -Endothelin-Rezeptorantagonisten - ET1-Antagonisten: z.B. Bosentan -Phophodiesterase-Hemmer (PDE 5 Inhibitoren): z.B. Sildenafil 3. Kausale Behandlung (ggf. Antikoagulation, ggf. Pulmonalis-Endarteriektomie, ggf. Behandlung der COPD oder andere Lungenerkrankungen |
|
|
Nichttuberkulöse Mykobakteriosen
|
MOTT (Mycobacteria other than tuberculosis)
Vorkommen in Böden und Wasser weltweit Infektion meist über Wasser Unterschiedliche Krankheitsbilder: 1. Tuberkuloseähnliche Lungenerkrankungen (M. kansasii, M. Avium, M. intracellulare) 2. Zervikale Lymphadenopathiem häufig im Kindesalter 3. Weichteil-, Knochen- und Hautinfektionen (M. marinum ->Schwimmbadgranulom) 4. Dissemierte Infektion bei AIDS-Patienten, meist bei T4<50/yl Th.: Entscheidung zur Therapie Nutzen-Risiko-Abwägung, lange Mehrfachtherapie notwendig |
|
|
Postnatale Toxoplasmose
|
Toxoplasma gondii = intrazellulär wachsendes Protozoon, Mensch ist Zwischenwirt, Katze Endwirt
Bradyzoiten persistieren lebenslang (bes. im ZNS), Reaktivierung bei Immunschwäche Immunkompetenter Patient: -meist chronisch latente Toxoplasmose ohne Symptome -1% der Infizierten: LK-Toxoplasmose mit LK-Schwellungen zervikal, nuchal, evtl. grippale Symptome, selten Uveitis, Hepatitis Immuninkompetenter Patient (AIDS): Schwerer Verlauf mit Hirntoxoplasmose, septische Streuung, interstitielle Pneumonie, Myokarditis, Augenbefall. Toxoplasmose verursacht die meisten ZNS-Infektionen AIDS-Kranker |
|
|
Konnatale Toxoplasmose
|
Bei der (relativ seltenen) frühen Fetusinfektion:
Abort oder schwerer Krankheitsverlauf: -Hepatosplenomegalie -Myokarditis -Interstitielle Pneumonie -Enzephalitis mit dem Trias Hydrozephalus, Chorioretinitis, intrazerebrale Verkalkungen Bei den häufigeren späten Fetusinfektionen: Asymptomatisch oder leichter Krankheitsverlauf |
|
|
Diagnose der Toxoplasmose
|
IgG: Sabin-Feldmann-Test
IgM PCR LK-Histologie (Piringer-Kuchinka-Lymphadenitis) |
|
|
Therapie der Toxoplasmose
|
Keine Behandlung von chronischen Toxoplasmose-Trägern. LK-Toxoplasmose heilt bei Immunkompetenten meist spontan ohne Therapie ab.
Therapieindikationen: -Schwerer Verlauf -Immunsuppression (z.B. AIDS) -Erstinfektion in der Schwangerschaft -Kongenitale Toxoplasmose Mittel der 1. Wahl: Pyrimethamin + Calciumfolinat + Sulfadiazin Bei AIDS-Patienten Primärprophylaxe mit Cotrimoxazol bei CD4<100-200/yl |
|
|
Übertragung der Toxoplasmose
|
-Genuss von zystenhaltigem rohem Fleisch (Mett!)
-Kontakt mit oozystenhaltigem Katzenkot, infizierter Gartenerde, Genuss von ungewaschenem Salat/ Gemüse |
|
|
Regelkreis Glukokortikoide
|
Hypothalamus -> CRH -> Produktion von ACTH im Hypophysenvorderlappen -> Stimulierung der NNR -> Kortisol
|
|
|
Pharmakologische Wirkungen von Glukokortikoiden
|
1. Glukoneogenese aus Aminosäuren und Intermediärprodukten -> Katabolismus mit Muskelatrophie und Osteoporose, diabetische Stoffwechsellage
2. Gesteigerter Fettabbau, Fettablagerung in der Leber -> Umverteilung des Fettes mit Stammfettsucht 3. Vermehrung der Leukozyten, Unterdrückung der B- und T-Zell-Aktivität -> Infektanfälligkeit, antiallergische Wirkung 4. Entzündungs-, Exsudations- und Proliferationshemmung -> antiphlogistische Wirkung, verzögerte Wundheilung 5. Hemmung der enteralen Calciumresorption, Förderung der renalen Calcium-Ausscheidung -> Hypokalzämie 6. Mineralkortikoide Wirkung -> Na-Retention, K-Ausscheidung |
|
|
Cushing-Schwellendosis
|
7,5mg Prednisolonäquivalent
|
|
|
Nebenwirkungen Kortisontherapie
|
-Fettzunahme am Stamm, Nacken, Gesicht, Fettverlust an Extremitäten
-Hypernatriämie, Wasserretention, Ödeme, Hypertonie -Diabetes mellitus -Infektanfällgkeit, Störung der Wundheilung -Steroidakne -erhöhtes Ulkusrisiko -Atrophie der Muskulatur, Steroidmyopathie (selten) -Osteoporose -Wachstumshemmung bei Kindern -Menstruationsstörungen bei Frauen -Psychische Störungen -Katarakt, Glaukom -Thromboseneigung |
|
|
Mit welchem Medikament kann man bei stattgehabter Tbc und geplanter immunsuppressiver Langzeittherapie eine Prophylaxe durchführen?
|
INH
|
|
|
Was gibt zusätzlich zu INH zur Prophylaxe der Neuropathie?
|
Vit B6
|
|
|
Ätiologie des Morbus Cushing
|
1. Exogen durch Langzeittherapie mit ACTH oder Glukokortikoiden
2. Endogene a) ACTH-abhängig -70% zentral: Mikroadenome des HVL (80%), primäre hypothalamische Überfunktion -Ektope (paraneoplastische) ACTH-Produktion -ektope CRH-Produktion (selten) -Alkohol-induziert b) ACTH-unabhängig -Kortisol-produzierende NNR-TU |
|
|
Morbus Cushing - Klinik
|
-Vollmondgesicht, Stiernacken, Stammfettsucht, supraklavikuläre Grube verstrichen
-Osteoporose -Myopathie (Pat. kann nicht aus Hockstellung ohne Arme aufstehen) -Diabetische Stoffwechsellage -Leukozytose, Erythrozytose, Thrombozytose -Hypertonie -Akne, Furunkulose, Striae rubrae, Atrophie der Haut -Frauen: Virilismus, Zyklusstörungen, Hirsutismus -Kinder: Wachstumsstillstand -evtl. psychische Veränderungen -selten Hypokaliämie Androgene Erscheinungen nur bei sekundären Cushing (ACTH-Erhöhung) oder bei Karzinomen der NNR |
|
|
Diagnose Morbus Cushing
|
-Niedrig dosierter Dexamethason-Hemmtest: Einnahme von 2mg Dexamethason um Mitternacht -> unzureichende Suppression des Serumkortisols am Folgetag 8:00 (>80mmol/l). Evtl. falsch positiv bei Pat. mit endogener Depression, unter Stress, bei Einnahme von hormonalen Antikoneptiva, Antiepileptika, bei Alkoholabusus
-Freies Kortisol im 24-Stunden-Urin erhöht -Aufhebung der zirkadianen Rhyhtmik der Serumkortisols im Tagesprofil Lokalisation: -CRH-Test (-> ACTH-Bestimmung) -hochdosierter Dexamethason-Hemmtest: 8mg Dexamethason um Mitternacht über 2 Tage -> Suppression des Serumkortisols um mindestens 50% bei zentralem Cushing, nicht aber bei adrenalem oder ektopem Cushing |
|
|
Welche 3 Formen des Morbus Cushing unterscheidet man grundsätzlich?
|
1. Zentrales Cushing (Hypothalamische Überfunktion oder HVL-Adenom
2. Adrenales Cushing (NNR-Tumor) 3. Paraneoplastisches Cushing (Ektope ACTH-Bildung) |
|
|
Neurokardiogene Synkope
|
Efferente Klassifizierung:
I Mischtyp (gleichzeitiger Abfall von art. RR und Hf, Hf fällt nicht länger als 10s unter 40bpm) II kardioinhibitorisch bei führender Bradykardie oder Asystolie (Hf <40bpm für 10s oder Asystolie >=3s; Typ A verausgegangener RR-Abfall, Typ B folgender RR-Abfall) III vasodepressorisch bei führender Hypotension (ausgepräger RR-Abfall bei max. Abfall der Hf um 10%) Afferente Klassifizierung: 1. Vasovagal (Orthostase, emotionaler Stress; tpyisch: Prodromi) 2. Carotis Sinus Synkope 3. Situational Syncope: Post-Exercise Synkope, Miktionssynkope, Hustensynkope, 4. Atypische Synkope (kein Auslöser erkennbar) |
|
|
Orthostatische Synkope
|
Efferente Klassifizierung
1. Klassische OH (RR-Abfall syst. >20mmHg oder diast. >10mmgH innerhalb von 3 Minuten) 2. Initiale OH (RR-Abfall >40mmHg syst. sofort nach dem Aufstehen, Normalisierung innerhalb von 30s) 3. Verzögerte OH (langsamer kontinuierlicher RR-Abfall, Fehlen der Bradykardie unterscheidet von Reflex-Synkope) 4. POTS (postural orthostatic tachycardia syndrome (Hf-Anstieg um 30bpm oder auf >120bpm, junge Frauen) Afferente Klassifizierung 1. Primary autonomic failure (z.B. M. Parkison) 2. Secondary autonomic failure (z.B. Diabetes mellitus, Urämie, Amyloidose) 3. Medikamenten/ Toxin-assoziierte Orthostase (Alkohol, Vasodilatatoren,etc.) 4. Volumenmangel (Blutung, Diarrhoe, etc.) |
|
|
Kardiale Synkope
|
1. Bradykardie
2. Tachkardie 3. Strukturelle Herzerkrankung |
|
|
Welche 3 Formen der Synkope unterscheidet man grundsätzlich?
|
1. Neurokardiogene Synkope
2. Orthostatische Snykope 3. Kardiale Synkope |
|
|
Therapie Morbus Cushing
|
Hormonell aktive NNR-Tumoren: Adrenalektomie, peri- und bis zu 2 Jahre postoperativ Substitution von Glukokortikoiden
Hypothalamisches-hypophysäres Cushing: Transnasales/transsphenoidaleoperative Adenomentfernung, bei KI evtl. Radioatio Paraneoplastische ACTH-Sekretion/ inoperabler NNR-Tu: Adrenostatische Substanzen (Ketoconazol + Octreotid, o-p-DDD, Aminoglutethimid, Metyrapon) |
|
|
Inzidentalome der Nebennieren
|
1. Hormonell inaktive Tumoren und Hyperplasien (60%)
2. Hormonell aktive Tumoren (30%): Phäochromozytom, Cushing-Adenom, Conn-Adenom 3. Nebennierenkarzinome (5%), Faustregel: Tumoren >6cm sind karzinomverdächtig, Tumoren <3cm wahrscheinlich gutartig 4. Metastasen (BC, Mamma-Ca, Nierenzell-Ca, GI-Ca, maligne Melanome) 5. Zysten, Pseudozysten, Hämatome, Myelolipome, Hämangiome, Tuberkulome) |
|
|
Welche hormonell aktiven Tumoren der Nebenniere gibt es und wie schließt man sie aus?
|
1. Phäochromozytom (Katecholamine im 24-Stunden-Urin)
2. Cushing-Adenom (niedrig dosierter Dexamethason-Hemmtest) 3. Conn-Adenom (Aldosteron/Renin-Quotient) |
|
|
Nebennierenrindenkarzinom
|
80% hormonell aktive Tumoren mit Zeichen der Hormonexzesses:
-Glukokortikoid-Exzess (Cushing) -Sexualsteroid-Exzess (Frauen: Virilisierung, Männer: Gynäkomastie) -Aldosteron-Exzess (Hypokaliämie, Hypertonie) Faustregel: Tumoren >6cm sind karzinomverdächtig, Tumoren <3cm sind wahrscheinlich gutartig Th.: Resektion adjuvant: Mitotane, ggf. Radioatio bei Fernmetastasen: Polychemotherapie |
|
|
Nebennierenrindeninsuffizienz
|
1. Primär (ACTH erhöht)
-80% Morbus Addison (Autoimmunadrenalitis, AK gegen 17-Alpha-Hydroxylase) -Karzinommetastasen -Infektionskrankheiten (CMV bei AIDS, Tbc) -akut: Waterhouse-Friderichsen-Syndrom = hämorrhagische Infarzierung infolge Meningokokkensepsis); Blutungen (Marcumar), fehlende Dosisanpassung bei Pat. mit NNR-Insuffizienz bei Infekten) 2. Sekundär (ACTH vermindert) -Insuffizienz von HVL oder Hypothalamus -Langzeitbehandlung mit Kortikoiden Bei der sekundären Form Aldosteron-Produktion nur wenig betroffen |
|
|
Klinik des Morbus Addison
|
4 Leitsymptome:
1. Schwäche und rasche Ermüdbarkeit 2. Pimentierung der Haut und Schleimhäute 3. Gewichtsverlust und Dehydratation 4. Niedriger art. Blutdruck außerdem: -abdominelle Beschwerden (Übelkeit, Durchfall, Obstipation, Schmerzen) -Verlust der Sekundärbehaarung der Frau (Androgenmangel) Addison-Krise: -Exsikkose, RR-Abfall, Schock -Pseudoperitonitis -Durchfälle, Erbrechen -Hypoglykämie, met. Azidose -Hypothermie, später Exsikkose-Fieber -Delir, Koma |
|
|
Diagnose des M. Addison/ der Nebennierenrindeninsuffizienz
|
-Vermindertes Natrium, erhöhtes Kalium (Na/K < 30)
-ACTH-Test: Serumkortisolbestimmung vor und 60 Minuten nach Gabe von ACTH (bei M. Addison oder länger bestehender sekundärer NNR-Insuff. Basalwert niedrig, kein Anstieg nach Gabe von ACTH) -Plasma ACTH (bei M.Addison erhöht, bei sek. NNR-Ins. erniedrigt, kein Anstieg nach CRH-Gabe) -NNR-Autoantikörper |
|
|
Therapie der NNR-Insuffizienz
|
Glukokortikoide, bei M. Addison zusätzlich Mineralkortikoide
-Hydrokortison 15-25mg, z.B. 10-5-5-mg -Fludrokortison (Astonin H), Dosis so, dass die Plasmareninaktivität im oberen Normbereich liegt (0,05-0,2mg/d) -evtl. zusätzlich DHEA (Dehydroepiandrosteron) bei Frauen, die über Libidoverlust klagen Bei allen Belastungen (Infekte, Operationen) muss die Dosis des Glukokortikoids erhöht werden (2-5faches der normalen Tagesdosis) Addison-Krise: -0,9% NaCl und 5% Glukose -100mg Hydrokortison als Bolus, dann 200mg/24h |
|
|
Polyglanduläre Autoimmunsyndrome
|
Typ I (Blizzard-Syndrom): Manifestation im Kindesalter, sehr selten
-Hypoparathyroidismus -M. Addison -Mukokutane Candidiasis -Primärer Hypogonadismus -Malabsorption -Autoimmunhepatitis Typ II (Carpenter-Syndrom): Manifestation im 3. Lebensjahrzehnt -M. Addison -Diabetes mellitus Typ I -Autoimmunthyreoiditis Hashimoto oder Basedow |
|
|
Phäochromozytom
|
-0,2% aller art. Hypertonien
-Katecholaminproduzierende neuroendokrine Tumoren des chromaffinen Gewebes des Nebennierenmarks oder der extraadrenalen Paraganglien. -85% gutartig, 15% maligne -90% einseitig, 10% doppelseitig -Sekretion von Adrenalin und Noradrenalin, extraadrenal gelegene P. nur NA, maligne P. auch Dopamin 25% hereditär im Rahmen von Syndromen (MEN 2, von Hippel-Lindau-S., M. Recklinghausen) -50% Paroxysmale art. Hypertonie, 50% Persistierende art. Hyptertonie |
|
|
Klinik des Phäochromozytoms
|
50% Paroxysmale art. Hypertonie
50% Persistierende art. Hyptertonie -Kopfschmerzen -Schwitzen -Blasswerden der Haut -Palpitationen -Gewichtsverlust (Hypermetabolismus) -Leukozytose -Hyperglykämie |
|
|
Diagnose des Phäochromozytoms
|
-Bestimmung der Katecholaminmetabolite Metanephrine und Normetanephrine im Plasma (strenge Abnahmebedingungen)
-Bestimmung der Katecholaminmetabolite im angesäuerten 24-Stunden-Urin -Bei Verdacht auf malignes Phäochromozytom zusätzlich Dopamin und Homovanillinmandelsäure -Clonidin-Hemmtest (bei Gesunden sinkt die Plasmakonzentration von Katecholaminmetaboliten) -Lokalisationsdiagnostik |
|
|
Therapie des Phäochromozytoms
|
-Laparoskopische oder oeprative TU-Entfernung in "No-touch-Technik"
-präoperative Alpha-Blockade und ggf. Betablockade (bei TAA) -Bei Inoperabilität Alphablocker (Phenoxybenzamin, Prazosin) Nach Operation >50% der Patienten normotensiv, bei den übrigen liegt zusätzlich eine essentielle Hypertonie vor. Im Langzeitverlauf 15% Rezidive. |
|
|
Evidenzklassen und Level
|
I Evidenz oder allgemeine Übereinkunft, dass eine Therapieform oder diagnostische Maßnahme effektiv, nützlich oder heilsam ist
II Widersprüchliche Evidenz/ unterschiedliche Meinungen A Empfehlung wird durch mindestens 2 randomisierte Studien gestützt B Empfehlung wird durch eine randomisierte Studie und/oder eine Metanalyse nicht-randomisierter Studien gestützt C Konsensus-Meinung von Experten |
|
|
Adrenogenitales Syndrom (AGS)
|
Autosomal rezessiv erbliche Störung der Cortisolsynthese in der NNR
90% der Fälle 21-Hydroxylase-Defekt -> aus den Hormonvorstufen wird vermindert Cortisol und Aldosteron gebildet, dafür mehr Androgene Klinik: 1. Virilisierung (Jungen:Hypogonadismus, verstärkte Entwicklung sekundärer Geschlechtsmerkmale (Pseudopubertas praecox; Mädchen: Pseudohermaphroditismus femininus) 2. Salzverlustsyndrom (niedriges Natrium, hohes Kalium, Erbrechen, Durchfall, Exsikkose - Fehldiagnose Pylorusstenose) Klassische Form mit Manifestation im Säuglingsalter Late-Onset (Pubertät) Cryptic forms Di.: Nachweis der Hormonvorstufen Th.: Lebenslange Behandlung mit Glukokortikoiden, ggf. Aldosteron, Antiandrogene |
|
|
Hypertrichose
Hirsutismus Virilismus |
Hypertrichose: Androgen-unabhängige Vermehrung der Behaarung am ganzen Körper
Hirsutismus: Abnorme Vermehrung der androgenabhängigen Behaarung vom männlichen Typ Virilismus: Hirsutismus + Vermännlichung der Stimme, der Körperproportionen, Klitorishypertrophie, Amenorrhoe |
|
|
Hypophysentumoren
|
ca. 10% aller Hirntumoren
40% endokrin inaktiv 60% endokrin aktiv: -Prolaktinom (häufigster) -Wachstumshormon prod. TU mit Akromegalie -ACTH-prod. TU mit zentralem Cushing Syndrom -TSH- und Gonadotropin-produzierende TU sind Raritäten |
|
|
Prolaktinom
|
70% Mikroporlaktinom (Prolaktin i.S. <200ng/ml, TU <1cm)
30% Makroprolaktinom (Prolaktin i.S. >200ng/ml, TU >1cm) Häufigster endokrin aktiver Hypophysentumor, w:m 5:1, Erkrankungsgipfel 3. und 4. Lebensdekade Kl.: Frauen: -Sekundäre Amenorrhoe, Sterilität, evtl. Osteoporose -Galaktorrhoe -Libidoverlust Männer: -Libido- und Potenzverlust -Gynäkomastie Beide: Zeichen der Raumforderung (Kopfschmerzen, Gesichtsfeldausfälle, HVL-Insuffizienz) Th.: 95% der Patienten sind mit Bromocriptin oder anderen Dopaminantagonisten behandelbar, darunter Normalisierung des Serumprolaktins und Rückbildung der Tumorgröße |
|
|
Welches sind die 4 hormonellen Achsen des HVL
|
1. GH (Growth Hormone) - Wachstumshormonachse
2. Gonadotropine (LH, FSH) - Gonadotrope Achse 3. TSH- Thyreotrope Achse 4. ACTH, MSH - corticotrope Achse Prolaktin |
|
|
Klinik der Hypophysenvorderlappeninsuffizienz
|
1. Ausfall GH im Wachstumsalter ->Kleinwuchs; beim Erwachsenen -> abdominale Fetteinlagerung, Reduktion der Muskelmasse, erhöhtes Arterioskleroserisiko, Osteoporose
2. Sekundärer Hypogonadismus -> Amenorrhoe, Libido- und Potenzverlust, Schwinden der Sekundärbehaarung, Depression 3. Sekundäre Hypothyreose -> Kälteintoleranz, Müdigkeit, bradykardie 4. Sekundäre NNR-Insuffizienz -> Adynamie, Gewichtsabnahme, wächserne Blässe durch Depigmentation (MSH), art. Hypotonie, Hypoglykämie; durch Glukokortikoidmangel SIADH 5. Ausfall von Prolaktin -> Agalaktie bei stillenden Frauen 7xA -Achselbehaarung (Gonadotr.) -Augenbrauenbehaarung (Gonadotr.) -Amenorrhoe (Gonadotr.) -Agalaktie (Prolaktin) -Apathie (TSH) -Adynamie (ACTH) -Alabasterfarbene Blässe (MSH) |
|
|
Ursachen für HVL-Insuffizienz
|
1. Hypophysenadenome, Kraniopharyngeome
2. Traumatisch, vaskulär (z.B. Sheehan-Syndrom = ischämische Hypophysennekreose durch größere peripartale Blutverluste) 3. Entzündlich-infiltrativ (granulomatöse Erkrankungen, Hämochromatose, Autoimmunhypophysitis) 4. selten hereditär |
|
|
Formen der HVL-Insuffizienz
|
1. Panhypopituitarismus (=Ausfall aller Funktionen = Morbus Simmonds)
2. Part. HVL-Insuffizienz |
|
|
Hypophysäres Koma
|
Unter Belastung (Trauma, Infekt) kann es durch ACTH-/ TSH-Mangel zum akuten hypophysären Koma kommen.
-Hypotonie, Bradykardie -Hypothermie -Hypoglykämie -Hypoventilation mit Hyperkapnie -Wächserne Blässe, Fehlen der Sekundärbehaarung |
|
|
Diabetes insipidus
|
Mangel an ADH oder vermindertes Ansprechen der Nieren auf ADH -> Konzentierungsfähigkeit der Niere herabgesetzt, vermehrte Ausscheidung eins verdünnten Urins.
1. Zentraler D.i. (idiopathisch, manchmal Autoantiokörper oder sekundär - Tumoren, Trauma, Meningitis) 2. Nephrogener D.i. (angeboren oder erworben bei Nierenerkrankungen) Trias: 1. Polyurie (5-25l/h) 2. Polydipsie 3. Asthenurie (verminderte Harnosmolalität) Di.: Durstversuch: Bei gesunden Personen kommt es zu einem Anstieg der Urinosmolalität, beim D.i. bleibt die Urinosmolalität <300mOsmol/l, Plasmaosmolalität steigt auf >295mOsmol an. Dann Testdosis ADH, beim zentralen D.i. steigt die Urinosmolalität an, beim renalen D.i. steigt sie nicht an. Th.: Zentr. D.i.: Desmopressin symptomatisch |
|
|
Berechnung der Serum-Osmolarität
|
2x Natrium + Harnstoff + Glukose
|
|
|
Hyponatriämie
|
Bei normaler Nierenfunktion meist Folge einer ADH-induzierten Reduktion der renalen Wasserausscheidung durch folgende Mechanismen:
1. Hypovolämiebedingte barorezeptorvermittelte ADH-Stimulation (z.B. dekompensierte Leberzirrhose, dekompensierte Herzinsuffizienz, Diarrhoe, Erbrechen, Verbrennungen, Peritonitis, Pankreatitis, Diuretikatherapie, Salzverlustniere, Aldosteronmangel 2. SIADH, z.B. durch Medikamente, paraneoplastisch, Legionellen-Pneumonie Lethargie, Orientierungsstörungen, Adynamie, Appetitstörungen Th.: Langsamer Ausgleich! |
|
|
Diabetische Polyneuropathie
|
Ursache möglicherweise Mikrozirkulationsstörungen der Vasa vasorum + metabolische Störungen
Auftreten meist beim unbehandelten oder schlecht eingestellten Diabetes mellitus -Frühzeichen: Erlöschen der ASR -sensible Reizerscheinungen, v.a. nachts! ("burning feet", Muskelkrämpfe im M. quadriceps und M. triceps surae, dumpfe oder lanzierende Schmerzen in der Lendengegend, in der Ilioinguinalregion, an der Vorderseite der Oberschenkel -Störung des Vibrationsempfindens, handschuh- und strumpfförmige Störung von Berührungs-, Temperatur- und Schmerzempfinden -selten: diabetische Amyotropie und diabetische Radikulopathie -diabetische Hirnnervenlähmungen (N. occulomotorius, N. abducens, N. fascialis -Autonome diabetische Neuropathie (kardiovaskulär, Magen-Darmtrakt, Urogenitaltrakt, Thermoregulation, neuroendokrines System) |
|
|
Autonome diabetische Neuropathie
|
1. Kardiovaskuläre ADN:
-verminderte Herzfrequenzvariabilität -Ruhetachykardie (Vagusschädigung) -asympathikotone orthostatische Hypotonie -inverse zirkadiane RR-Rhythmik 2. ADN des GIT -Gastroparese -postprandiale Diarrhoe und Obstipation im Wechsel -anorektale Dysfunktion -Ösophagusmotilitätsstörungen 3. ADN des Urogenitalsystems -Blasenenteerungsstörungen -erektile Impotenz 4. ADN des neuroendokrinen Systems -Reduktion der hormonellen Gegenregulation bei Hypoglykämie (verminderte Wahrnehmung!) 5. ADN der Thermoregulation -verminderte Schweißsekretion -Vasodilatation (warmer und trockener diabetischer Fuß) 6. ADN der Pupillen |
|
|
PNP bei Urämie
|
25% der Patienten mit Urämie/ Dialyse
-sensible Reizerscheinungen ("burning feet") -leichte Paresen |
|
|
Critical Illness PNP
|
Endogen toxische PNP bei Patienten mit langandauernder Sepsis und MOV
-überwiegend motorische Nerven befallen ->schwere, schlaffe, atrophische Lähmungen aller Extremitäten einschließlich der Atemmuskulatur -sensible Fasern nicht/ kaum betroffen Günstige Spontanprognose |
|
|
Mangelerscheinungen bei radikaler vegetarischer Ernährung
|
1. Eisen (Plummer-Vinson-Syndrom mit Zungenbrennen und schmerzhafter Dysphagie, Cheilitis angularis, Anämie)
2. Vitamin B-Komplex -Vit B1, Thiamin (->Beriberi - symmetrische sensomotorische PNP) B2, Riboflavin; Niacin (Pellagra: Dermatitis - Casal Kragen, Diarrhoe, Demenz; PNP) B 6 B12 (->megaloblastäre Anämie, funikuläre Myelose mit Paraparese der Beine und sensibler Ataxie, Hunter-Glossitis, psychische Veränderungen) 3. Calcium bei streng veganer Ernährung (-> Osteoporose) 4. Jod |
|
|
Wieviel Kohlenhydrate enhält eine BE
|
12g
Für 1 BE werden beim Typ I Diabetes ca. 1-2 IE Insulin benötigt |
|
|
Thiaminmangel
|
Vit B1
-Häufigste Ursache: Alkoholismus -Malabsorption -Lebererkrankungen (verminderte Aktivierung zu Thiamindipshosphat) -Diuretikatherapie (->vermehrte Ausscheidung) Kl.: -Leistungsabfall, Müdigkeit, Kopfschmerzen -Beriberi: PNP, Anorexie, Ataxie, Apathie, Schwäche, Parästhesien, Nystagmus, Ophthalmoplegie; Ödeme, Herzversagen -Wernicke-Korsakow-Syndrom: Desorientiertheit, Ataxie, Nystagmus Bei Alkoholikern immer vor Glukosegabe 100mg Thiamin |
|
|
WHO Klassifikation des Diabetes mellitus
|
I Typ 1 Diabetes: meist immunologisch bedingte Zerstörung der Beta-Zellen; Sonderform: LADA (latent autoimmune diabetes in adults)
II Typ 2 Diabetes (Inulinresistenz, sekretorischer Defekt der Beta-Zellen, Apoptose der Beta-Zellen) III Andere Diabetesformen A MODY (Maturity Onset Diabetes of the Young - autosomal dominant vererbter Defekt der Beta-Zellfunktion) B Genetische Defekte der Insulinwirkung C Erkrankungen des exokrinen Pankreas D Endokrinopahtien (Akromegalie, Cushing Syndrom, Phäochromozytom, Hyperthyreose, u.a.) E Medikamentös induzierter Diabetes (Kortikoide, SD-Hormone, Thiazide, hormonelle Kontrazeptiva) F Infektionen (CMV, kongenitale Röteln) G seltene immunolog. Formen H Genetische Snydrome (Down, Klinefelter, Turner) IV Gestationsdiabetes |
|
|
Vaskuläre hämorrhagische Diathesen
|
1. Hereditär:
-Morbus Osler Rendu-Weber (autosomal dominant; punktförmige Teleangiektasien bes. Zunge, Lippen, Nasenschleimhaut; rezidivierende GIT-Blutungen, evtl. arterioven. Malformation in der Lunge -> Hämoptoe und im Gehirn). Teleangiektasien verschwinden auf Spateldruck. 2. erworben -Lanzeitbehandlung mit Kortison -Vitamin C Mangel -Purpura Schoenlein Hennoch |
|
|
Skorbut
|
Vitamin C Mangel
-Schleimhautblutungen -Wadenschmerzen -follikuläre Hyperkeratose -Blutungen in Muskulatur und unter Periost -hypochrome Anämie -psychische Veränderungen (Gleichgültigkeit, Depression) -Müdigkeit -Gingivitis -Katarakt -Infektneigung -begünstigt Karzinomentstehung |
|
|
Schweregradeinteilung des Diabets mellitus
|
IGT impaired glucose tolerance
NIR non-insulin-requiering IRC insulin requiering for control IRS insulin requiering for survival |
|
|
Diabetes mellitus Typ I - Ätiologie
|
20% der Typ-I-Diabetiker haben eine positive Familienanamnese
>90% HLA DR3 und/oder DR4 Nachweis von Auto-AK: -ICA (Inselzell-AK) sehr aufwändig -Anti-GAD-AK (GADA) (Glutamatdecarboxylase) -Anti-IA-2-AK (IA-2A) (Tyrosinphosphatase 2) GADA und IA-2A zusammen >90% der Typ I Diabetiker |
|
|
Diabetes Typ II
|
-Störung der frühen postprandialen Insulinsekretion
-Apoptose der Inselzellen -Insulinresistenz Metabolisches Syndrom: Zunächst Insulinresistenz -> erhöhte Insulinspiegel -> vermehrtes Hungergefühl -> Adipositas Hohe Insulinspiegel vermindern Sensibilität und Dichte der Insulinrezeptoren (Circulus vitiosus) |
|
|
Metabolisches Sydrom
|
Zusammentreffen der 4 Risikofaktoren
1. Stammbetonte (viszerale) Adipositas 2. Dyslipoproteinämie (hohe TG, niedriges HDL) 3. essentielle Hypertonie 4. Glukosetoleranzstörung/ Diabetes Typ II IDF-Definition (International Diabetes Federation): Abdominelle Adipositas (m Taillenumfang >=94cm w >=80cm) plus 2 der folgenden Faktoren: -TG>150mg/dl -HDL <50mg/dl wm <40mg/dl m -RR >130/85mmHg -Nüchtern-Glukose >100mg/dl oder Typ II Diabetes |
|
|
Diabetes mellitus und Vererbung
|
Typ I:
Risiko bei Erkrankung des Vater 5%, der Mutter 2,5%, beide 20%, eineiige Zwillinge 35% Typ II Ein Elternteil 50%, eineiige Zwillinge 100% |
|
|
Klinik des Diabetes mellitus
|
-Müdigkeit, Leistungsminderung
-Polyurie, Polydipsie, Gewichtsverlust -Nächtliche Wadenkrämpfe, Sehstörungen (Elektrolytverschiebungen) -Haut: Pruritus, Furunkulose, Candida, Rubeosis diabetica, Necrobiosis lipoidica, Granuloma anulare, Bullosis diabeticorum) -Amenorrhoe, Potenzstörungen |
|
|
Komplikationen des Diabetes mellitus
|
1. Makroangiopathie
-KHK mit diffusem Verteilungsmuster und häufigem Hauptstammbefall; gestörte Anginawahrnehmung -pAVK -AVK der Hirnarterien 2. Mikroangiopathie -Glomerulosklerose (M. Kimmelstiel-Wilson) bei Typ I oder unspez. Veränderungen bei Typ II ->(Mikro-) Albuminurie, Niereninsuffizienz -diabetische Retinopathie (proliferativ oder nicht-proliferativ) -diabetische Neuropathie -diabetisches Fußsyndrom 3. Diabetische Kardiomyopathie 4. Neigung zu Haut- und Harnwegsinfekten 5. Fettstoffwechselstörungen 6. Fettleber 7. Coma diabeticum 8. Hyporeninämischer Hypoaldosteronimus (Hyperkaliämie, Hyponatriämie) |
|
|
Mikroalbuminurie
|
30-300mg/24h oder 20-200mg/l im Spontanurin
<200mg/dl Mikroalbuminurie >200mg/dl Makroalbuminurie |
|
|
Wirkungen von Aldosteron, ADH, natriuretischen Peptiden
|
Aldosteron: Natrium- und Wasserretention; ANP hemmt Aldosteron, Angiotensin II stimuliert Aldosteron-Sekretion, Reninsekretion durch verminderte Dehnung der Barorezeptoren der juxtaglomerulären Zellen der Niere)
ADH: Harnkonzentrierung, Ausscheidung von Natrium, Retention von Wasser. ADH steuert die Wasserbilanz des Körpers! Gauer-Henry-Reflex: erhöhte Vorhofdehnung vermindert ADH-Freisetzung; Osmorezeptoren im Hypothalamus Natriuretische Peptide hemmen RAAS ANP (atriales natriuretisches Peptid, Vorhof) BNP (brain natriuretic peptide, Ventrikel) CNP (Typ C natriuretisches Peptid, Blutgefäße) |
|
|
Wodurch wird der Natrium-Wasserhaushalt geregelt?
|
1. ADH
2. RAAS 3. ANP, BNP, CNP |
|
|
Diabetisches Fußsyndrom
|
Schweregradeinteilung nach Wagner (Grad 0-5;
Tiefe der Läsion) und Armstrong (A-D, begünstigende Faktoren) 1. Neuropathischer diabetischer Fuß: -warme, trockene Haut, Hyperkeratosen -tastbare Fußpulse, Dopplerindex normal -Störung der Abrollbewegung -ggf. Malum perforans an druckbelasteten Stellen 2. Diabetisch-neuropathische Osteoarthropathie mit Nekrosen im Bereich der Metatarsalgelenke und Tarsometatarsalgelenke, Charcot-Fuß 3. Ischämischer Fuß bei pAVK -kühl, blass bis livide -Nekrosen, Gangrän -Fußpulse nicht tastbar, verminderter Dopplerindex, verminderte transkutante O2-Sättigung |
|
|
Normaler Dopplerindex
|
Knöchel - Arm >0,9
|
|
|
Diagnose des Diabetes mellitus
|
Plasmawerte entscheidend!
Nüchtern-Glukose >= 126mg/dl: Diabetes mellitus 100-125mg/dl: Abnorme Nüchtern-Glukose <100mg/dl normal Gelegenheitszucker >= 200mg/dl und Symptone: Diabetes mellitus OGTT 2h-Wert >= 200mg/dl: Diabetes mellitus 2h-Wert 140-199mg/dl gestörte Glukose-Toleranz 2h-Wert < 140mg/dl normal |
|
|
Thiazide
|
Blockierung des Na-Cl-Cotransportes am frühdistalen Tubulus -> 15% des filtrierten Natriums werden ausgeschieden (und Kalium)
HCT (Esidrix) Xipamid (Aquaphor) NW: - Hyponatriämie, Hypokaliämie, Hyperkalzämie -RR-Senkung, Thrombosegefahr -Stoffwechselstörungen: Erhöhung von Glukose, Harnsäure, LDL und TG -Aktivierung des RAAS Cave Digitalisintoxikation |
|
|
Schleifendiuretika
|
Blockierung des Na/K-2Cl -Carriers im aufsteigenden Teil der Henle´schen Schleife
-> Ausscheidung von 40% des filtrierten Natriums Furosemid führt außerdem zu einer direkten Venodilatation, wodurch es noch vor Eintritt der diuretischen Wirkung die Vorlast senkt. Bei Gabe von NSAR ist die Wirkung von Diuretika vermindert. NW.: - Hyponatriämie, Hypokaliämie, Hypokalzämie -Aktivierung des RAAS -RR-Abfall, Thromboseneigung -Hörverlust |
|
|
sequentielle Nephronblockade
|
Bei Diuretikaresistenz unter Schleifendiuretika nicht ständig höher dosieren sondern ein Thiazid hinzugeben -> sequentielle Nephronblockade -> Diurese nimmt wieder zu.
Resistenz gegen Schleifendiuretika wird durch kompensatorische Resorptionssteigerung im distalen Tubulus verursacht. Dort greifen Thiazide an. |
|
|
Kalium-sparende Diuretika
|
Hemmung der Na-Absorption und K-Sekretion im Sammelrohr
Nur mäßige diuretische Wirkung, keine Monotherapie, Kombination mit Thiaziden 1. Aldosteronantagonisten Spironolacton (Aldactone) Eplerenon (Inspra) (hemmt nur Mineralkortikoidrezeptoren, weniger hormonelle NW, teuer) 2. Aldosteron-unabhängige K-sparende D: Amilorid Triamteren |
|
|
Normale Nierenschwelle für Glukose
|
ca. 180mg/dl
|
|
|
OGTT
|
Oraler Glukose Toleranz-Test
75g Zucker, BZ-Messung nach 120 Minuten -> BZ>= 200mg/dl: Diabetes mellitus -> BZ 140-199mg/dl: gestörte Glukosetoleranz |
|
|
HbA1c
|
Glyciertes Hämoglobin, "Blutzuckergedächtnis" für die letzten 8 Wochen, Referenzbereich <6,2%
|
|
|
Gewichtsklassifikation (Adipositas)
|
BMI (kg/m2)
18,5-24,9 Normalgewicht 25-29,9 Übergewicht (Präadipositas) 30-34,9 Adipositas I 35-39,9 Adipositas II >= 40 Adipositas III |
|
|
Energiegehalt der Nährstoffe
|
1g Kohlenhydrat = 4,1kcal
1g Eiweiß =4,1 kcal 1g Fett = 9,3 kcal 1g Alkohol = 7,1 kcal |
|
|
Täglicher Energiebedarf eines Erwachsenen
|
Normalgewicht x 32 bei leichter körperlicher Arbeit
Normalgewicht x 40 bei mittelschwerer körperlicher Arbeit Normalgewicht x 48 bei schwerer körperlicher Arbeit |
|
|
Zusammensetzung gesunder Kost
|
% der Gesamtkalorien
Eiweiß 10-15%; bei diabetischer Nephropathie eiweißarme Diät Fett 25-30%, dabei hoher Anteil ungesättigter Fettsäuren (max. 10-15% gesättigte FS) |
|
|
Omega 3/6 FS
|
Omega 3 (Alpha-Linolensäure)
Omega 6 (Linolsäure) essentielle, mehrfach ungesättigte FS (mehrere Doppelbindungen) Nahrungsquellen: -Fisch (Hering, Ölsardinen) -Nüsse -Samenöle, z.T auch in tierischen Produkten (je nach Fütterung) |
|
|
Einfach ungesättigte FS
|
z.B. Ölsäure (pflanzliche Öle:
Fettsäuren mit einer Doppelbindung Olivenöl, Rapsöl, Nussöle, bestimmte Sonnenblumenöle) Palmitoleinsäure (Fisch, Samenöle) |
|
|
Phasengerechte Stufentherapie des Typ-II-Diabetes
|
1. Gewichtsnormalisierung
2. Orales Antidiabetikum (Metformin bei Übergewichtigen 1. Wahl) 3. Metformin + 2. OAD 4. Basalinsulin + OAD 5. Basalinsulin + Bolusinsulin +OAD 6. Konventionelle oder intensivierte Insulintherapie |
|
|
Orale Antidiabetika
|
A Nicht-insulinotrop
1. Biguanide (Metformin) 2. Alpha-Glukosidase-Hemmer (Acarbose, Miglitol) 3. Glitazone (Pioglitazon, Rosiglitazon) B insulinotrop 1. Sulfonylharnstoffe (Glibenclamid, Glimepirid) 2. Sulfonylharnstoffanaloga (Glinide) 3. DPP-4-Inhibitoren (Sitagliptin - Januvia) 4. Inkretinmimetika (Exanitide -Byetta) |
|
|
Biguanide
|
Metformin
-verzögerte Glukoseresorption aus dem Darm -Hemmung der hepatischen Glukoneogenese -verstärkte Glukoseaufnahme in die Muskulatur -Appetitsenkender Effekt First line Therapie für übergewichtige Typ II Diabetiker Cave Niereninsuffizienz, Herzinsuff., resp. Insuff. |
|
|
Alpha-Glukosidashemmer
|
Acarbose
Kompetetive Hemmung der Glukoamylase, Saccharase, Maltase in der Dünndarmmukosa -> Abflachung der postprandialen BZ-Peaks NW: Bauchschmerzen, Flatulenz, Diarrhoe |
|
|
Glitazone
|
Pioglitazon
Rosiglitazon Insulin-Sensitizer, verbessert Empfindlichkeit der peripheren Zellen für Insulin NW: Gewichtszunahme, Verschlechterung einer Herzinsuffizienz, erhöhtes Frakturrisiko bei Frauen, Erhöhung der Leberenzyme Kombinationstherapie mit Metformin oder Sulfonylharnstoffen |
|
|
Sulfonylharnstoffe
|
Glibenclamid
Glimepirid Stimulation der Insulinsekretion NW: Hypoglykämie! (erhöhtes Risiko bei Niereninsuffizienz, Alkoholkonsum, körperl. Anstrengung) Sekundärversagen nach ca. 10 Jahren aufgrund einer Erschöpfung der Beta-Zellen |
|
|
Sulfonylharnstoffanaloga
|
Glinide: Repaglinide (Novonorm), Nateglinide (Starlix)
Postprandiale Stimulation der Insulinsekretion Hypoglykämierisiko niedriger als bei Sulfonylharnstoffen |
|
|
DPP-4-Inhibitoren und Inkretinmimetika
|
GLP-1 (glucagon-like-peptide 1) wird mahlzeitenabhängig von den neuroendokrinen L-Zellen des Dünndarms aus Proglukagon gebildet und innerhalb weniger Minuten vom Enzym Dipeptidyl-Peptidase-4 inaktiviert.
1. DPP-4-Inhibitoren (Sitagliptin, Vildagliptin) -> Hemmung der DPP-4 -> Erhöhung des GLP-1-Spiegels ->Stimulierung der Insulinsekretion und Hemmung der Glukagon-Sekretion 2. Inkretinmemetika (Exanitide): GLP-1 Analoga |
|
|
Wie kommt es bei Diabetikern zur Bildung von Ketonkörpern?
|
Glukosemangel in den Zellen des Fettdepots -> Fettsäuren werden nicht zu Triglyceriden synthetisiert sondern verlassen so das Fettgewebe
-> in der Leber Abbau zu Ketonkörpern über Actyl-CoA -> Ketonkörper vermindern Permeabilität der Zellen für Glukose |
|
|
Insulinbedarf des Menschen
|
Tagesbedarf 40IE (bei Normalgewicht)
20IE für Nahrungsaufnahme 20IE für basalen Stoffwechselbedarf 1IE Insulin senkt den BZ um 30-40mg/dl (bei Pat. ohne Insulinresistenz) 1KE (10g KH) erhöht den BZ um 30-40mg/dl |
|
|
Welche Arten von Insulin gibt es?
|
A Kurz wirksame Insuline
1. Normalinsulin: Wirkeintritt nach 30-60Minuten, Wirkdauer ca. 5 Stunden; z.B. Actrapid 2. Kurz wirksame Insulinanaloga: Wirkeintritt nach 10Minunten, Wirkdauer ca. 3,5h; Humalog, Novorapid B Verzögerungsinsuline 1. Intermediärinsulin: NPH-Insuline - Neutrale-Protamin-Hagedorn-Insuline: Wirkeintritt nach 60 Minten, Wirkdauer 9-18 Stunden; Insuman Basal 2. Lanzeitinsuline: Wirkeintritt nach 60 Minuten, Wirkdauer bis 24h; Lantus, Levemir C Mischinsuline Actraphane 30 (30% Normalinsulin) Humalog Mix 25 (25% Insulin Lispro) Alle NPH-Insuline müssen mindestens 10x geschwenkt werden! |
|
|
Verschiedene Formen der Insulintherapie
|
1. Konventionelle IT:
Mischinsuline, 2/3 - 3/4 der Tagesdosis morgens vor dem Frühstück, Rest vor dem Abendessen. 2. Intensivierte Insulintherapie -Basis-Bolus: Zirkadiane Insulinempfindlichkeit, Insulinbedarf pro KH 2IE-1IE-1,5IE -Insulinpumpe |
|
|
Ursachen für morgendliche Hyperglykämie
|
1. Somogyi-Effekt: Insulindosis abends zu hoch, nächtliche Hypoglykämie mit reaktiver morgendlicher Hyperglykämie
2. Dawn-Phänomen: Erhöhter Insulinbedarf in der 2. Nachthälfte durch vermehrte nächtliche Wachstumshormonsekretion (bes. Typ I Diabetes) |
|
|
Behandlungsziele beim Diabetes mellitus
|
-BZ nüchtern 80-110mg/dl, postprandial <140mg/dl
-LDL<100mg/dl/ <70mg/dl HDL >45mg/dl TG <150mg/dl -HbA1c Typ I normnah, Typ II <6,5/ 7 (ACCORD-Studie, ADVANCE-Studie) -RR <130/80mmHg, bei Proteinurie <125/75mg/dl (ACE-Hemmer, AT1-Blocker) Rauchverbot Bei persistierende Proteinurie Proteinrestriktion, <6g/d NaCl) Diabetiker mit mindestens einem weiteren Risikofaktor prophylaktisch ASS 100 |
|
|
Glasgow Coma Scale
|
Öffnen der Augen
4 spontan 3 auf Ansprache 2 auf Schmerzreiz 1 fehlt Verbale Reaktion 5 orientiert 4 verwirrt 3 einzelne Worte 2 Laute 1 fehlt Motorische Antwort 6 folgt Aufforderungen 5 gezielte Schmerzreaktion 4 ungezielte Schmerzreaktion 3 Beugesnergismen 2 Strecksynergismen 1 fehlt |
|
|
Hyperglyämisches Koma
|
1. Ketoazidotisches Koma (Typ I)
2. Hyperosmolares Koma (Typ II) -Volumenmangel, Schock (durch Hyperosmolarität) -Nierenversagen (durch Hypovolämie) -Pseudoperitonimus (peritoneale Reizerscheinungen, Magen-Darm-Atonie, Magenüberblähung) -bei Ketoazidose evtl. Kussmaul-Atmung |
|
|
Bewusstseinsstörungen
|
1. Somnolenz: Pat. schläft oft ein, erscheint verlangsamt, kann aber durch einfache äußere Reize zu adäquaten Reaktionen veranlasst werden.
2. Sopor: Pat. kann nur durch starke Reize vorübergehend und unvollständig geweckt werden. 3. Koma: Vollständig Unerweckbarkeit |
|
|
Therapie bei Hyperglykämischem Koma
|
1. Volumen
2. Kalium 3. Insulin (BZ <50mg/dl/h senken, zunächst nicht <250mg/dl; cave Retinaschäden) 4. Azidose nur vorsichtig behandeln |
|
|
Ursachen für Hypoglykämien
|
1. Beim Diabetiker:
-Überdosierung von Insulin oder Sulfonylharnstoffen -verminderte Nahrungszufuhr -Alkohol -körperl. Belastung Beim Nicht-Diabetiker: -Insulinom -schwere Lebererkrankung (verminderte Glukoneogenese) -Urämie (vermindertes Substrat für Glukoneogenese) -NNR- oder HVL-Insuffizienz -selten paraneoplastisch (Sekrektion insulinähnlicher Peptide) -Dumping-Syndrom (Spät-Dumping) |
|
|
Sonographie der Schilddrüse
|
Länge 4-7cm
Breite 1-3cm Dicke 1-2cm Volumen = Länge x Breite x Tiefe Männer <25ml Frauen <20ml -Knotenstruma: echogleich oder echoreich -Adenome: Autonome Adenome (mikrofollikuläre Adenome) meist echoarm mit echoarmem Randsaum; Makrofollikuläre Adenome reflexreich, meist kalte Knoten -Zysten, Fibrosen, Verkalkungen (regressive Veränderungen), Verkalkung auch bei Ca Morbus Basedow: Pralle Schwellung der Drüse über den Isthmus hinweg, diffus fleckige Echoarmut Hashimoto: Kleine atrophische SD |
|
|
Euthyreote Struma
|
Vergrößerung der Schilddrüse bei normaler Hormonproduktion
30% der deutschen Bevölkerung betroffen Komplikationen: -Tracheale Kompikationen bis zur Tracheomalazie -Entwicklung einer SD-Autonomie -Entwicklung kalter Knoten Th.: -Jodidsubstitution (200ygd) -Kombinationstherapie aus T4 und Jodid (einschleichende Dosierung, Beginn mit 50yg, Steigerung bis ca. 100yg) Ziel: Niedrig normales TSH, normales T3 und T4 |
|
|
Strumagrade nach WHO
|
Grad 0: nur sonographisch feststellber (Männer >25ml, Frauen >20ml)
Grad 1: tastbar aber nicht sichtbar Grad 2: sichtbar und tastbar |
|
|
Hyperthyreose
|
1. Immunogen (M. Basedow mit oder ohne Struma)
2. Schilddrüsenautonomie (unifokal, multifokal oder disseminiert) 3. iatrogen (Amiodaron, Hyperthyreosis factitia) Klinik: -Struma -psychomotorische Unruhe (Tremor, Schlafstörungen) -Sinustachykardie, TAA, systolische Hypertonie -Gewichtsverlust trotz Heißhunger -warme, feuchte Haut -weiches, dünnes Haar -Wärmeintoleranz, Schweißausbrüche -Durchfall -Osteoporose -Myopathie -pathologische Glukosetoleranz -Fettleber -Zyklusstörungen, Infertilität Merseburg-Trias: Struma, Exophthalmus, Tachykardie Bei älteren Patienten häufig oligosymptomatischer Verlauf |
|
|
Diagnostik und Therapie bei Hyperthyreose
|
Di.:
-TSH-Rezeptor-AK (TRAK) verursachen M. Basedow und sind erhöht, außerdem Erhöhung von Anti-TPO-AK -Sono, Szinti Th.: -Schwefelhaltige Thyreostatika (Dosisverhältnis von Carbimazol : Thiamazol = 1,5:1), hemmen Synthese aber nicht Inkretion, 6-8 Tage Latenz bis Wirkeintritt -Perchlorate (Irenat): hemmen Aufnahme von Jodid in die SD-Zellen, rasche Blockierung bei Notwendigkeit der Gabe von jodhaltigen KM Bei M. Basedow 1 Jahr Therapie (50% Rezidive danach) -operativ -Radiojodtherapie |
|
|
Thyreotoxische Krise
|
3 Stadien nach Hermann:
I Tachykardie oder TAA, Fieber bis 41°C, Schwitzen, Exsikkose, psychomotorische Unruhe, Angst, Erbrechen, Durchfälle, Muskelschwäche II Zusätzlich Bewusstseinsstörungen, Somnolenz, Psychose III Koma, evtl. NNR-Insuff., Kreislaufversagen Th.: -Thiamazol 80mg i.v. alle 8h -Kaliumperchlorat (1500mg/d) -ggf. Plasmapherese bei iodinduzierter Hyperthyreose, SD-Resektion -Volumen, Kalorien, Elyte -Beta-Blocker -Glukokortikoide (hemmen die Konversion von T3 zu T4) |
|
|
Endokrine Orbitopathie
|
>90% mit M. Basedow assoziiert, TSH-Rezeptoren finden sich auch im Orbitagewebe.
Infiltration mit autoreaktiven T-Lymphozyten, Fibroblastenvermehrung, Einlagerung von Glykosaminoglykanen in periorbitalem Gewebe und äußeren Augenmuskeln -Stellwag-Zeichen: Seltener Lidschlag -Darymple-Zeichen: sichtbarer Sklerastreifen oberhalb der Hornhaut beim Blick geradeaus -Graefe-Zeichen: Zurückbleiben des Oberlides bei Blicksenkung -Möbius-Zeichen: Konvergenzschwäche -Frühzeichen: Schwellung der lateralen Augenbrauenpartie -Lichtscheu, Fremdkörpergefühl, schmerzhafter Druck hinter den Augen, Visusverschlechterung |
|
|
Schweregrade der endokrinen Orbitopathie
|
nach Grußendorf und Horster:
I Anamnestisch Beschwerden: Fremdkörpergefühl, Tränen, Lichtscheu, retrobulbäres Druckgefühl II Lidretraktion und BG-Veränderungen (Konjunktivitis, Chemosis, periorbitale Schwellung) III Protrusio bulbi IV Augenmuskelblockaden mit Doppelbildern V Hornhautulcerationen VI Visusverlust bis Erblindung |
|
|
Therapie der endokrinen Orbitopathie
|
-Euthyreose, Hypothyreose vermeiden!
-Verzicht auf Rauchen -Lokale Therapie (getönte Augengläser, Dexpanthenolsalbe) -Kortikoide -Retrobulbärbestrahlung der Orbita -Operative Dekompression der Orbita 30% Besserung 60% keine Änderung 10% Verschlechterung |
|
|
Subakute granulomatöse Thyreoiditis de Quervain
|
oft im Anschluss an Luftwegsinfekt
Kl.: -Abgeschlagenheit, Krankheitsgefühl, Fieber -Schilddrüse druckschmerzhaft -hohe BSG! normale Leukos -zunächst oft hyperthyreot, später wieder euthyreot -Sono: "landkartenartige" Schilddrüse DD: Akute eitrige Thyreoiditis Th.: Spontanheilung in 80% der Fälle, keine Thyreostatika -NSAR, Glukokortikoide (diagnostisch!) |
|
|
Extrahepatische Komplikationen der
Hepatitis C |
-Kryoglobulinämie
-Glomerulonephritis -Hashimoto-Thyreoiditis -Sjögren-Syndrom -Porphyria cutanea tarda -idiopathische Thrombozytopenie |
|
|
Schilddrüsenkarzinom
|
1. Differenziert
a) papillär (lymphogene Met.) 60% b) follikuläre (hämatogene Met.) 30% 2. Anaplastisch (5%) 3. Medullär (C-Zell) (5%) |
|
|
Akute intermittierende hepatische Porphyrie
|
Aktivitätsminderung der Porphobilinogen-Desaminase (PGB-D) -> Absinken des hepatischen Häm-Pools -> Steigerung der D-ALS-Synthase -> Anstieg der Häm-Präkursoren (ALS, PBG, Porphyrine)
Manifestation durch Stress, Infektion, Trauma, Operation, Fasten, Hpoglykämie, Alkohol, zahlreiche Medikamente (z.B. MCP, Diazepam, Metamizol) Kl.: 1. Abdominalschmerzen 2. neurologische/ psychiatrische Symptome, periphere Lähmungen 3. kardiovaskuläre Symptome (Hypertonie, Tachykardie) -SIADH -rötlicher, beim Stehenlassen nachdunkelnder Urin; Hoesch-Test, dunkle Flecken in der Unterwäsche Th.: Hämarginat und Glukose evtl. Intervallprophylaxe mit Hämarginat |
|
|
Porphyria cutanea tarda
|
= chronische hepatische Porphyrie, häufigste Prophyrie
Mangel an Uroporphyrinogen-Decarboxylase in der Leber; angeboren (50% autosomal dominant, 50% sporadisch; oder erworben (Hep. C, Alkoholabusus, AIDS, Hämodialyse) Kl.: -Fotodermatose mit erhöhter Vulnerabilität, Hyperpigmentierung, Blasenbildung -dunkler Urin -Leberschäden mit Porphyrineinlagerungen, sonographisch gelegentlich multiple echoreiche Rundherde Th.: -Meidung auslösender Noxen -Therapie einer Hep. C -Aderlässe (-> Verminderung des Lebereisens) -Chloroquin (-> bildet mit Porphyrinen Komplexe, die mit dem Urin ausgeschieden werden.) -Lichtschutz |
|
|
Porphyrien PG
|
Störungen der Biosynthese des Häm, das in 8 enzymatischen Schritten aus Glycin und SuccinylCoA gebildet wird
Nach dem Ort der hauptsächlichen Enzymproduktion unterscheidet man erythropoetische und hepatische Porphyrien. |
|
|
Pathogenese der Hyperurikämie
|
A Primäre Formen
99% der Fälle Störung der tubulären Harnsäuresekretion, polygen vererbt, Manifestation bei purinreicher Ernährung und Übergewicht 1% Überproduktion von Harnsäure -Lesch-Nyhan-Syndrom (Trias Hyperurikämie, progressive Niereninsuffizienz, Neigung zur Selbstverstümmelung) -Kelley-Seegmiller-Syndrom (Trias Hyperurikämie, Nierensteine, neurolog. Störungen) B Sekundäre Formen 1. Vermehrte Harnsäurebildung bei erhöhtem Nukleinsäuren-Turnover bei Leukämien, hämoly. Anämien, Tumoren unter Therapie 2. Verminderte renale Ausscheidung bei Niereninsuffizienz, Laktazidosen, Ketoazidosen, Schleifendiuretika, Thiazide |
|
|
Gicht - Klinik
|
4 Stadien:
I Asymptomatische Hyperurikämie II Akuter Gichtanfall III Interkritisches Stadium (symptomloses Intervall zwischen 2 Gichtanfällen) IV Chronische Gicht mit Tophusbildungen und irreversiblen Gelenkveränderungen Akuter Gichtanfall: Podagra (Großzehengrundgelenk), Sprunggelenk/ Fußwurzel (15%), Kniegelenk (Gonagra, 10%), Zehengelenke, Fingergelenke, bes. Daumengrundgelenk (Chiragra), Handgelenk, Ellenbogengelenk ->begleitet von allgemeinen Entzündungszeichen (Fieber, Leukozytose), Hyperurikämie im Anfall nicht obligat Chronische Gicht: -Tophi (Uratablagerungen) in Weichteilen (Ohrmuschel, Schleimbeutel, Sehnenscheiden) oder Knochen -Renale Manifestationen (Uratnephrolithiasis, Uratnephropathie - primär abakterielle interstitielle Nephritis), selten akutes Nierenversagen |
|
|
Therapie bei Hyperurikämie
|
1. Allgemeinmaßnahmen: Körpergewicht normalisieren, viel trinken, purinarme Diät, sparsamer Alkoholgenuss
Cave Diuretika (Schleifendiuretika und Thiazide vermindern die Harnsäureausscheidung.) 2. Medikamentös: a) Akuter Anfall: NSAR, evtl. Kortikoide Colchicin Mittel 2. Wahl wegen NW b) Dauerbehandlung (Harnsäure>9mg/dl oder manifeste Gicht): -Allopurinol (Urikostatikum, Hemmung der Xanthinoxidase); initial Auslösung von Gichtanfällen möglich, cave Allopurinol-Hypersensitivitätssyndrom -Rasburicase (Fasturtec) bei Tumorlysesyndrom (rekombinante Uratoxidase) -Benzbromaron und Probenecid (Urikosurika) bei Allergie gegen Allopurinol, einschleichend dosieren,viel trinken, cave Harnsteinbildung |
|
|
Metabolisches Syndrom
|
IDF 2005 (International Diabetes foundation)
Abdominelle Adipositas (Männer >= 94cm, Frauen >= 88cm) plus 2 der folgenden Faktoren: -Hypertonus >130/85mmHg) -Triglyceride > 150mg/dl -HDL Männer <40mg/dl, Frauen <50mg/dl -Nüchtern-Gluocse > 100mg/dl oder Typ II Diabetes |
|
|
NCEP-ATP-III-Empfehlungen zum LDL-Cholesterin
|
0-1 Risikofaktor LDL < 160mg/dl
>= 2 RF LDL <130mg/dl KHK, PORCAM-Score >20% LDL <100mg/dl Bei Diabetes und hohem Risiko evtl. <70mg/dl |
|
|
Schutzwirkung von HDL
|
-Cholesterinrücktransport aus den peripheren Geweben
-antiinflammatorische Wirkung -anti-oxidative Wirkung -anti-thrombotische Wirkung -Stimulation der NO-Synthase -Inhibition des Tissue-factors |
|
|
Klinische Einteilung der Dyslipidämien
|
Einteilung nach Fredrikson nicht mehr gebräuchlich
1. LDL-Hypercholesterinämie (evtl. LDL-Rezeptordefekt) 2. Hypertriglyceridämie 3. gemischte Hyperlipoproteinämie (Erhöhung LDL, Erhöhung Triglyceride, Verminderung HDL) Prototyp diabetische Dyslipoproteinämie 4. Isolierte HDL-Erniedrigung |
|
|
Sekundäre Ursachen für Dyslipidämie
|
-Übergewicht, Inaktivität, kohlenhydratreiche Ernährung, Alkohol, Rauchen
-Diabetes mellitus -Nephrotisches Syndrom, chronische Niereninsuffizienz -Hypothyreose -Cholestase -Medikamente (Kortikoide, HIV-Proteaseinhibitoren, Beta-Blocker, Thiazide, Östrogene, Androgene, Ciclosporin A) |
|
|
Medikamte zur Behandlung von Dyslipproteinämien
|
1. Statine (HMG-CoA-Reduktase-Hemmer, CSE-Hemmer): Einnahme abends, weil die Aktivität der HMG-CoA-Reduktase nachts am höchsten ist. -> Senkung des LDL
2. Cholesterinresorptionshemmer (Ezetimib) ->Hemmung der Resorption von biliärem und alimentärem Cholesterin aus dem Darm 3. Fibrate (z.B. Bezafibrat) -> komplexe Wirkung, unter anderem gesteigerter Katabolismus triglyceridreicher Lipoproteine 4. Nikotinsäure (Niacin) -> Hemmung der Lipolyse im peripheren Fettgewebe; NW Flush 5. Gallensäureaustauschharze (z.B. Cholestyramin) 6. Omega-3-FS |
|
|
Vorgehen bei LDL-Hypercholesterinämie
|
1. Lebensstiländerung, Kontrolle der Werte nach 2-3 Monaten
2. Statin 3. Bei unzureichendem Ansprechen: -Dosissteigerung -stärker wirksames Statin (Atorvastatin, Rosuvastatin) -Kombinationstherapie mit Ezetimib -ggf. Kombinationstherapie mit Anionenaustauscherharzen |
|
|
Vorgehen bei Hypertriglyceridämie
|
1. Lebensstilmodifikation
2. Fibrat oder Niacin oder Omega-3-FS, bei nachgewiesener Atherosklerose +Statin |
|
|
Typen des HI-Virus
|
HIV I (häufigster Typ weltweit)
3 Hauptgruppen (M, N , O) M Subtypen A-K HIV II, 6 Subtypen |
|
|
CDC-Stadienteinteilung der HIV-Infektion
|
Klinische Kategorien:
A Akutes retrovirales Syndrom oder asymptomatisch oder LAS B Symptomatisch aber nicht A oder C C AIDS-Indikator-Krankheiten Bereiche der T4-Zellen 1 >500/yl 2 200-499/yl 3 <200/yl |
|
|
AIDS Kategorie A
|
1. Akute HIV-Krankheit (30%): 1-6 Wochen nach Erstinfektion Mononukleose-ähnliches Krankheitsbild mit Fieber, LK-Schwellungen, Splenomegalie, Angina, gel. Exantham, Myalgien
2. LAS (40%): Generalisierte Lymphadenopathie: >3 Monate LK-Schwellungenan mindestens 2 extrainguinalen Stellen, Fehlen von Allgemeinsymptomen, evtl. seborrhoische Dermatitis 3. Asymptomatische Infektion (Latenzphase) |
|
|
AIDS Kategorie B
|
Nicht-AIDS-definierende Krankheiten, die durch Immundefekt begünstigt werden:
-subfebrile Temperaturen -chronische Diarrhoe (>1Monat) -zervikale Dysplasie, Carcinioma in situ -periphere Neuropathie -Listeriose -Candidose > 1 Monat -Herpes Zoster, Befall mehrerer Dermatome, Gefahr intraokulärer Komplikationen -orale Haarleukoplakie |
|
|
AIDS - Kategorie C (AIDS - definierende Krankheiten)
|
1. Wasting-Syndrom
2. HIV-assoziierte Enzephalopathie 3. Opportunistische Infektionen, die AIDS definieren 4. Malignome, die AIDS definieren |
|
|
Opportunistische Infektionen, die AIDS definieren
|
-Zerebrale Toxoplasmose
-Pneumocystis jiroveci Pneumonie -Candidose von Bronchien, Lunge, Trachea oder Ösophagus -Kryptokokkose -Histoplasmose -Kokzidiomykose -rezidivierende bakterielle Pneumonien -atypische Mykobakteriose -Tuberkulose -Salmonellen-Sepsis -CMV (Erblindung!) -Herpes simplex -Progressive multifokale Leukenzephalopathie (PML - JC-Virus-Infektion) |
|
|
Malignome, die AIDS definieren
|
-Kaposi-Sarkom
-Non-Hodgkin-Lymphome (z.B. Burkitt-Lymphom) -Invasives Cervixkarzinom |
|
|
Wasting Syndrom (AIDS)
|
->ungewollter Verlust von > 10% des Körpergewichtes und chronische Diarrhoe
Vorkommen bei ca. 14% der unbehandelten Patienten, CD4<200/yl |
|
|
HIV-assoziierte Enzephalopathie (HIVE)
|
Infektion der Mikroglia mit koneskutiver Zerstörung des ZNS
Vorkommen bei 15-20% der unbehandelten Patienten, CD4<200/yl Klinik: Subkortikale, langsam fortschreitende Demenz mit kognitiven (Konzentrations- und Gedächtnisstörungen), motorischen (Gangstörung, Feinmotorik-Störung), emotionalen (Depression) und selten vegetativen (Miktionsstörungen) Symptomen |
|
|
Zerebrale Toxoplasmose als AIDS definierende Erkrankung
|
Kl.: Fieber, Verwirrtheit, Psychosyndrome, Kopfschmerzen, zerebrale Krampfanfälle
MRT: Intrazerebrale Abzesse mit KM-Enhancement Th.: -Clindmycin oder -Sulfadiazin + Pyrimethamin + Folinsäure oder -Atovaquon Primärprophylaxe: Cotrimoxazol |
|
|
Pneumocystis jiroveci Pneumonie als AIDS definierende Erkrankung
|
Häufigste Pneumonieform bei AIDS-Patienten
Kl.: -Belastungsdyspnoe -trockener Reizhusten -subfebrile Temperaturen -Hypoxie -meist LHD erhöht Meist unaufällig Auskultation der Lunge, im Rö bilaterale milchglasartige Infiltrate Th.: -Cotrimoxazol, bei Hypoxie gleichzeitg Steroide -Mittel 2. Wahl: Pentamidin i.v. Primärprophylaxe/ Sekundäprophylaxe: -Cotrimoxazol -2.Wahl: Pentamidin-Inhalation |
|
|
Atypische Mykobakteriose als
AIDS-definierende Erkrankung |
Meist Mykobakterium avis oder intrazellulare
Kl: -Fieber -Gewichtsverlust -Bauchschmerzen -Erhöhung der AP -Hepatosplenomegalie Di: -Blutkultur -Kultur von Punktaten -Kultur von respiratorischen und gastrointestinalen Sekreten Th: Ethambutol + Clarithromycin + Rifabutin |
|
|
Tuberkulose als AIDS definierende Erkrankung
|
Betont in den Unterfeldern ohne Kavernenbildung, gehäuft miliare Verläufe
Tuberkulin-Hauttest oft negativ Cave bei der Therapie Interaktionen mit antiretroviraler Therapie |
|
|
CMV als AIDS-definierende Erkrankung
|
Kl.:
-gastrointestinaler Befall -retinaler Befall -Pneumonie -Enzephalitis Th.: -Valganciclovir oder Ganciclovir |
|
|
Klinik der konnatalen HIV-Infektion
|
-Frühgeburtlichkeit
-Dystrophie -Kraniofasziale Dysmorphie -ZNS-Schäden: Kortikale Atropie und Verkalkung der Stammganglien mit Ataxie -Opportunistische Infektionen (Pneumocystis jirovecii, Haemophilus influenzae, Candidosen, CMV-Infekte, Herpes-Virus-Infekte -Lymphoide interstitielle Pneumonie mit chronischem Verlauf (LIP) |
|
|
Antiretrovirale Therapie - Indikation
|
1. Symptomatische HIV-Infektion
2. Asymptomatische HIV-Infektion -CD4<350/yl -CD4>=500/yl und Zusatzkriterien -> Einzelfallentscheidung -HIV-RNA>100.000 Kopien/ml ->engmaschige Kontrollen mit CD4 -CD4 350-500/yl und Zusatzkriterien -> Behandlungempfehlung Zusatzkriterien: -HIV-RNA>100.000/yl -Schwangerschaft -Alter >50J -HCV- oder HBV-Koinfektion -hohes kardiovaskuläres Risiko -Absinken der CD4-Zellen |
|
|
Antiretrovirale Therapie - Substanzklassen
|
1a. NRTI (Nukleosidische Reverse-Transkriptase-Hemmer)
z.B. Zidovudin (AZT), Stavudin (d4T), Lamivudin (3TC), Abacavir (ABC), Emtricitabin (FTC) 1b. NtRTI (Nukleotid-analoge Reverse Transkriptase-Inhibitoren) z.B. Tenofovir (TDF) 2. NNRTI (Nicht-nukleosidische Reverse-Transkriptase-Hemmer) z.B. Nevirapin (NVP), Efavirenz (EFV), Etravirin (ETV) 3. PI (Protease-Inhibitoren) z.B. Saquinavir (SQV), Nelvinavir (NFV), Indinavir (IDV), Ritonavir (RTV), Darunavir (DRV), usw. 4. Fusionsinhibitor (Enfuvirtide - T-20) 5. CCR5-Inhibitor (Maraviroc - MCV) 6. Integrase-Inhibitor (Raltegravir - RAL) |
|
|
HAART
|
Highly active antiretroviral therapy
Kombinationstherapie mit mindestens 3 antiretroviralen Substanzen In der Regel 2 NRTI, 1 NNRTI oder 1 PI NNRTI und PI haben ein breites Spektrum an Interaktionen (Metabolismus über Cytochrom P 450) Langzeitnebenwirkungen: kardiovaskuläre Erkrankungen, Lipodystrophiesyndrom Lebenslange Therapie, bei Compliance von 95% Ansprechrate >80% Monitoring über CD4-Zellzahl und Viruslast |
|
|
Antiretrovirale Postexpositionsprophylaxe
|
Innerhalb von 2 Stunden -> Reduktion des Infektionsrisikos um 80%
3er Kombination für 4 Wochen (2 NRTI + 1 PI) |
|
|
Häufigste Erreger einer Reisediarrhoe
|
-40% ETEC (Enterotoxinbildende E. coli)
-5-10% Salmonellen -5-10% Campylobacter jejuni -5-10% Shigellen |
|
|
Verschiedene klinische Verlaufsformen von infektiösen Durchfallerkrankungen
|
1. Dysenterische Durchfälle: Kolikartige Bauchschmerzen, Diarhhoe mit Beimischung von Blut/ Schleim/ Eiter
a) Typus Amöbenruhr (Entamoeba histolytica) -> Symptomatik entwickelt sich über längere Zeit, anfallsartiger verlauf mit beschwerdeärmeren Intervallen b) Typus bakterielle Ruhr (Shigellen, EHEC, EIEC) -> akut oder perakut einsetzende Symptomatik 2. Nichtdysenterische Druchfälle: Akut einsetzende mildere Symptomatik a) Typus enterotoxische Form (ETEC, Salmonellen, Enteroviren, Vibrio cholerae, "Lebensmittelvergiftung") -> akut einsetzende Symptomatik, evtl. mit Erbrechen b) Typus Resorptionsstörungen (Giardia lamblia) -> Faezes schaumig und voluminös mit gelegentlichen Beimengungen von Fett und unverdauter Nahrung |
|
|
Antibiotische Therapie von infektiösen Durchfallerkrankungen
|
Indikation für Antibiotika:
-blutige Durchfälle -schwerer Krankheitsverlauf -Fieber Mittel der Wahl: Ciprofloxacin, Alternative: Cotrim Bei Amöbiasis und Lambliasis: Metronidazol Clostridien: Metronidazol, Vanco |
|
|
EHEC-Infektion
|
Enterohämorrhagische E. coli
Shigatoxin Häufigkeitsgipfel bei Kleinkindern Übertragung fäkal-oral oder über den Genuss von EHEC-haltigen Tierprodukten (Rohmilch, rohes Rindfleisch) Verlaufsformen: Asymptomatisch, wässrige Durchfälle, blutig-wässrige Durchfälle Postinfektiös mögl. HUS (Hämolytisch-urämisches Syndrom), 50% der Kinder werden dialysepflichtig Therapie: Symptomatisch |
|
|
Einteilung der Salmonellosen
|
O-Ag: Oberflächen-Antigen
H-Ag (Geißel-Antigen) Vi-Ag (Virulanzantigen) Durch Bestimmung der O und H-Antigene mit Antiseren Unterteilung in 2000 Serotypen nach dem Kauffmann-White-Schema |
|
|
Salmonellen-Dauerausscheider
|
10 Wochen nach Krankheitsbeginn noch Salmonellen im Stuhl
v.a. bei Salmonella typhi 2 Typen: -Galleausscheider (2/3) Versuch der Sanierung mit Ciprofloxacin 1 Monat -Dünndarmausscheider (1/3) Salmonellen-Dauerausscheider haben ein erhöhtes Risiko für Gallengangskarzinom |
|
|
Salmonellen Gastroenteritis
|
Brechdurchfälle durch Endotoxine von S. enteritidis oder S. typhimurium
Bei Immunschwäche Gefahr der Salmonellensepsis (Absiedlungen an Endokard, Pleura, Meningen, Knochen, Gelenken) Dauerausscheider sehr selten. Salmonellen sind mehrere Monate überlebensfähig, werden durch Einfrieren nicht abgetötet. Nur bei schwerem Verlauf antibiotische Therapie (Ciprofloxacin). |
|
|
Campylobacter-Enterokolitis
|
In Europa häufigste durch Lebensmittel übertragene bakterielle Diarrhoe
Kl.: -Kopf- und Gliederschmerzen -Fieber -wässrige, oft auch blutige Diarrhoe mit kolikartigen Bauchschmerzen 10% Rezidive Ko.: -Reaktive Arthritis -sehr selten Guillain-Barre-Syndrom |
|
|
Lebensmittelvergiftung
|
Ursache: Enterotoxin-bildende Bakterien (meist Staph. aureus)
Kurze Inkubationszeit, Übelkeit, Erbrechen, Diarrhoe Cave: Botulismus! |
|
|
Shigellose
|
Bakterielle Ruhr
Erkrankung der Not- und Kriegszeiten, Unhygiene, Resistenzminderung Bei leichtem Verlauf wässrige Diarrhoe, bei schwerem Verlauf blutig-schleimig-eitrige Durchfälle, Darmkrämpfe, Tenesmen Ko.: Reaktive Arthritis, HUS |
|
|
Amöbiasis
|
Entamoeba histolytica sensu stricto, Tropen/ Subtropen
2 Entwicklungsstadien: 1. Zyste (werden von Infizierten mit dem Stuhl ausgeschieden, können monatelang infektiös bleiben) 2. Vegetatives Stadium: Aus den Zysten bilden sich im Kolon Trophozoiten (Minutaformen), nach Phagozytose von Erythrozyten Magnaformen Kl.: 1. Amöbenkolitis: Himbeergeleeartige Durchfälle (Schleimfäden und Blutspuren), ausgestanzte Ulcerationen der Kolonschleimhaut - "Flaschenhals-Ulkus"; chronischer Verlauf in Form einer rezidivierenden Kolitis möglich 2. Leberabszesse; nur in 30% ist anamnestisch eine Kolitis vorausgegangen Th.: Metronidazol, Sanierung durch Gabe eines Kontaktamöbizids (z.B. Paromomycin) |
|
|
Yersiniose
|
Yersinia enterocolica
Yersinia pseudotuberculosis (Osteuropa, Russland) Erregerreservoir im Tierreich (Infektion durch Milchprodukte, rohes Fleisch) Kl: -Gastroenteritis -Pseudoappendizitische Verlaufsform: Akute Lymphadenitis mesenterica und Ileitis terminalis (DD Appendizitis) -Enterokolitische Verlaufsform (1-2 Wochen Durchfall, kolikartige UB-Schmerzen, gel. chronischer Durchfall) Ko.: -Reaktive Arthritis -Erythema nodosum (HLA-B27) |
|
|
Kryptosporidiose
|
Cryptosporidium parvum, obligat intrazellulär lebendes Protozoon
Bei immunkompetenten Personen asymptomatische Infektion oder selbstlimitierender Verlauf Häufige Durchfallerkrankung bei AIDS-Patienten, ggf. mit Befall der Gallengänge, bronchopulmonale Infektion Di.: Mikroskopischer Nachweis von Kryptosporidien oder Oozysten im Stuhl Keine sicher wirksame Therapie verfügbar, in Erprobung Nitrazoxamid |
|
|
Botulismus
|
Nahrungsmittelvergiftung durch Neurotoxine von Clostridium botulinum
Sporen können unter Luftabschluss aufkeimen und Toxine bilden, z.B. in eingemachten verunreinigten Lebensmitteln Außerdem: Säuglingsbotulismus, Wundbotulismus Botulismustoxin hemmt irreversibel die Acetylcholinfreisetzung in den peripheren cholinergen Nervenendplatten Kl.: -Übelkeit, Erbrechen -Periphere Lähmungen: Zuerst Hirnnerven: weite Pupillen, Doppelbilder, Ptosis, Dysarthrie, Dysphagie; dann Fortschreiten nach kaudal bis zur Atemlähmung Th.: Antitoxin vom Pferd |
|
|
Clostridium difficile Infektionen
|
Grampositive Stäbchen mit Sporenbildung
Hypervirulente Stämme: Ribotypen 027 und 078 Bildung von Toxin A (Enterotoxin -> vermehrte Sekretion von Elektrolyten und Flüssigkeit) und Toxin B (Zytotoxin, das die Mukosa schädigt) CDAD (Clostridien assoziierte Diarrhoe) PMC (pseudomembranöse Kolitis) Toxisches Megakolon Th: CDAD: Metronidazol für 10 Tage 1. Rezidiv: Metronidazol für 10 Tage 2. Rezidiv: Metronidazol oder Vancomycin 14-21 Tage Schwere Fälle: Kombinationstherapie |
|
|
Cestoden (Bandwürmer)
|
1. Taenia saginata (Rinderbandwurm)
2. Taenia solium (Schweinebandwurm) 3. Diphylobothrium latum (Fischbandwurm) 4. Echinococcus granulosus (Hundebandwurm) 5. Echinococcus multilocularis (Fuchsbandwurm) |
|
|
Nematoden (Fadenwürmer)
|
1.Ascaris lumbricoides (Spulwurm
->evtl. Lungeninfiltrat, Ileus, allergische Hautreaktion) 2. Trichuris trichiura (Peitschenwurm -> Eier wie Zitronen, mögl. hämorrhagische Kolitis) 3. Enterobius vermicularis (Madenwurm -> Analer Juckreiz, Appendizitis, KLebstreifen) 4. Ancylostoma duodenale, Necator americanus (Hakenwurm -> Löffler Lungeninfiltrat, abdom. Schmerzen) 5. Strongyloides stercoralis (Zwergfadenwurm -> Dermatitis, Bronchitis, Enterokolitis) 6. Trichinella spiralis (Trichinose ->enterale Phase -> extraintestinale Phase (Fieber, Gesichtsödem, Muskelschmerzen und -schwäche, Lungenpassage, ZNS) |
|
|
Protozoen im Darm
|
1. Entamaeba histolytica (Himbeergeleeartige Durchfälle, Tenesmen; Leberabszesse)
2. Giardia lamblia (Durchfall, chronischer Verlauf mit Malresorption und Gewichtsverlust möglich) |
|
|
Filarien
|
Wucheria Brugia (Stechmücken)
Loa (Fliegen) Onchocerca (Kriebelmücken) 1. Lymphatische Filariasis: Lymphödeme, Elephantiasis 2. Onchozerkose: Chronische Dermatitis, Keratitis -> Flussblindheit 3. Loiasis: Juckende Schwellungen der Haut, schmerzhafte subkonjunktivale Durchwanderung des Auges |
|
|
Keuchhusten
|
Bordetella pertussis
3 Stadien: 1. St. catarrhale (1-2 Wochen) 2. St. convulsivum (Stakkatohusten, Erbrechen; 4-6 Wochen) 3. St. decrementi (6-10 Wochen) Ko.: -Durch den Husten: subkonjunktivale Blutungen, Synkopen, Apnoe bei Säuglingen, Leistenhernien, Rippenfrakturen, Pneumothorax -Pneumonie, Sinusitis, Otitis media Leukozytose mit rel. Lymphozytose Th.: Makrolide |
|
|
Coxsackie-Virusinfektion
|
Coxsackie A:
-Herpangina Zahorsky mit Myalgien, Cephalgien, Fieber und Bläschen am weichen Gaumen, der Uvula und den Tonsillen -Sommergrippe -Lymphozytäre Meningitis -Hand-Fuß-Mund-Krankheit Coxsackie B: -Bornholm-Krankheit (Pleurodynie mit gürtelförmigen, stechenden Schmerzen im Bereich des Thorax/ Oberbauches) -Myokarditis, Perimyokarditis -Lymphozytäre Meningitis -Sommergrippe |
|
|
Mumps
|
Parotitis epidemica
Paramyxovirus parotitidis Kl.: Schmerzhafte Schwellung der Parotis, in 75% der Fälle bds. mit abstehenden Ohrläppchen und Schmerzen beim Kauen Ko.: -Pankreatitis -Orchitis (25% der Männer), Oophoritis (5% der Frauen) -bei 10% Meningitis (mit guter Prognose), 0,1% Meningoenzephalitis mit ernster Prognose -0,01% Innenohrschwerhörigkeit Therapie symptomatisch, bei kompliziertem Verlauf Immunglobulin |
|
|
Diphtherie
|
Corynebacterium diphtheriae (keulenförmiges grampositives Stäbchen), Diphtherietoxin A (aktiv toxisch) und B (bindend)
Kl.: 1. Lokalinfektion -Rachendiphtherie: Angina mit festhaftenden weißen Belägen, die beim Abstreifen bluten (Pseudomembranen) -Kehlkopfdiphtherie mit Erstickungsgefahr (Croup: Bellender Husten) -süßlicher Geruch nach vergärenden Äpfeln -blutiger Schnupfen bei Säuglinden -Wunddiphtherie 2. Systemische Intoxikation 4-5 Tage nach Lokalinfektion: Hohes Fieber, Erbrechen. Ko.: Ödematöse Halsschwellung (Cäsarenhals), Früh- oder Spätmyokarditis, PNP mit Paresen der motorischen Kopfnerven, Schluckstörungen, selten Nierenschäden Th.: Antitoxin! Penicillin oder Erythromycin |
|
|
Listeriose
|
Listeria monocytogenes
Gefährdet sind Pat. mit Immunsuppression und Schwangere/ Feten/ Neugeborene Infektion durch Rohmilchprodukte, Rohwürste, geräucherten Fisch, Salat Postnatale Listeriose: Grippeähnlich; bei Immunsuppression: Meningoenzphalitis, bei Leberzirrhose Peritonitis, hämatogene Streuung in verschiedene Organe, "Pseudo-Pfeiffer" Neonatale Listeriose: -Frühinfektion -> Früh- oder Totgeburt, Sepsis, Atemnotsyndrom, Hautläsionen (Granulomatosis infantoseptica) in der ersten Lebenswoche -Spätinfektion ->Meningitis, Granulomatosis infantoseptica in der 2. Lebenswoche Th.: Amoxicillin + Aminoglykosid; Cephalosporine unwirksam |
|
|
durch Zecken übertragbare Krankheiten
|
1. Borreliose
2. Frühsommermeningo-enzephalitis 3. Humane granulozytäre Ehrlichose |
|
|
Lyme-Borreliose
|
Borrelia burgdorferi, übertragen durch Zecken
5-35% der Zecken tragen den Erreger in Endemiegebieten, Infektionsrate 10%, Erkrankungsrate 1% St. I (1-6 Wochen): Erythema migrans St. II (bis 6 Monate): Lymphozytäre Meningoreadikulitis Bannwarth evtl. mit Faszialisparese, Myokarditis evtl. mit AV-Block, Arthritis (v.a. Knie-/ Ellenbogengelenk) St. III (>6 Monate bis Jahre): Acrodermatitis chronica atrophicans, Polyneuropathie, Enzephalopathie Th.: St. I: Doxycyclin oder Amoxicillin St. II: Ceftriaxon i.v. |
|
|
FSME
|
Frühsommer-Meningo-Enzephalitis
Übertragung durch Zecken, 5% der Zecken Träger in Endemiegebieten FSME:L-Borreliose 1:100-300 Kl.: 70-90% asymptomatisch Symptomatischer Verlauf: 1. Fieber mit grippalen Symptomen 2. nach Fieber-freiem Intervall erneuter Fieberanstieg mit Meningitis oder Meningoenzephalitis 10% Defektheilungen 1% Letalität Aktive Immunisierung möglich |
|
|
Häufigste Erreger einer bakteriellen Meningitis
|
Säuglinge: E.coli, B-Streptokokken, Listerien
Kleinkinder: Haemophilus influenzae, Meningokokken, Pneumokokken Erwachsene: Pneumokokken, Meningokokken, Listerien Pat. mit Immunsuppression: Listerien, Cryptococcus neoformans, M. tuberculosis |
|
|
Meningismuszeichen
|
-Nackensteifigkeit
-Kernig -Lasegue -Brudzinski |
|
|
Rheumatoide Arthritis - Pathomechanismus
|
Synovialitis -> Arthritis, Bursitis, Tendovaginitis
Interaktion von Lymphozyten und Monozyten mit Produktion von proinflammatorischen Zytokinen (IL1, IL6, IL15, TNF-Alpha), Immunglobulinen und Autoantikörpern gegen das Fc-Fragment des IgG (Rheumafaktor). Aktivierung von Komplement und Freisetzung von Entzündungsmediatoren sowie knorpelaggressiven Enzymen (Kollagenase, Elastase). Pannus: Verdickung der Synovialis durch Invasion makrophagenähnlicher Zellen (Typ A-Synoviozyten) und Proliferation fibrolastenähnlicher Zellen (Typ-B-Synoviozyten). Beteiligung innerer Organe durch Immunkomplexvaskulitis. |
|
|
Rheumatoride Arthritis - Epidemiologie
|
1% aller Erwachsenen
2% aller >55 J w:m 3:1 HLA DR4 mit schwerem Krankheitsverlauf assoziiert |
|
|
Klinik der rheumatoiden Arthritis
|
1. Unspez. Allgemeinsymtome
2. Polyarthritis, Tendovaginitis, Bursitis; meist symmetrischer Beginn an den kleinen Gelenken (Fingergrundgelenke und proximale Interphalagealgelenke); schmerzhafter Händedruck (Gaensslen-Zeichen); Karpaltunnelsyndrom, Baker-Zyste 3. Rheumaknoten (in Sehnen subkuten) 4. Nagelveränderungen 5. Sicca-Syndrom in 30% der Fälle 6. Organmanifestationen: -Perikarditis, Myokarditis, Klappenläsionen -Pleuritis, Lungenfibrose -Leberenzymerhöhungen -selten fokal membranöse GN -Keratokonjunktivitis sicca -vorzeitige Aterosklerose |
|
|
Karpaltunnelsyndrom
|
Kompression des N. medianus unter dem Lig. carpi transversum
Parästhesien von Daumen, Zeige- und Mittelfinger; Schmerzen im Kompressionsbereich v.a. nachts Phalen Zeichen: Auslösung der Beschwerden durch Beugung oder Streckung im Handgelenk Hoffmann-Tinel-Test: Auslösen der Beschwerden durch Beklopfen des Karpaltunnels DD: -Rheumatoide Arthritis -Überlastung -fehlverheilte distale Radiusfraktur -Hypothyreose, Akromegalie, Diabetes mellitus, Schwangerschaft -Gicht -Amyloidose -idiopathisch |
|
|
Sonderformen der rheumatoide Arthritis
|
1. Caplan-Syndrom: RA + Silikose
2. Felty-Syndrom: Schwere Verlaufsform der RA im Erwachsenenalter (Hepatosplenomegalie, LK-Schwellungen, Granulopenie) 3. Alters-RA = LORA (Late onset rheumatoid arthritis), >60.LJ, oft aggressiver Verlauf 4. Juvenile idiopathische Arthritis (JIA) <16.LJ -Morbus Still (systemisch mit hohem Fieber, Exanthem, Polyserositis, Hepatosplenomegalie, LK-Schwellungen -weitere Formen der Poly- oder Oligoarthritis im Kindesalter 5. RS3PE-Syndrom: Remitting seronegative symmetric synovitis and pitting edema; symmetrisch teigige Schwellung des Handrückens |
|
|
Komplikationen der rheumatoiden Arthritis
|
1. Funktionsverlust und Fehlstellung von Gelenken (Schwanenhalsdeformität, Knopflochdeformität, ulnare Deviation, atlantoaxiale Subluxation)
2. NW der antirheumatischen Therapie (Magen-/Duodenalulzera, Analgetikanephropathie, Infektionen unter Immunsuppressiva, Osteoporose) 3. Sekundäre Amyloidose vom AA-Typ mit nephrotischem Syndrom 4. Gehäuftes Auftreten maligner Erkrankungen 5. Koronare Herzkrankheit |
|
|
Laborparameter bei der rheumatoiden Arthritis
|
1. Unspezifische Entzündungszeichen = Aktivitätszeichen (BSG, CRP)
2. Immunologische Befunde: -Rheumafaktor - Autoantikörper gegen körpereigenes IgG, Fc-Fragment (initial bei 40% positiv, im Verlauf bei 80%) -Anti-CCP (cyclisches citrulliniertes Peptid) (hohe Spezifität -MCV-Ak (mutiertes citrulliniertes Vimentin) höhere Sensitivität bei gleicher Spezifität wie CCP-Ak |
|
|
Bildgebende Verfahren bei der Diagnose einer rheumatoide Arthritis
|
-Arthrosonographie
-KM-MRT (Inflammation, Knorpel- und Knochenerosionen werden bis zu 2 Jahre früher erkannt als im konvent. Röntgen) -Röntgen der Hände, Vorfüße, HWS -Weichteilszintigraphie |
|
|
Röntgenstadien der rheumatoiden Arthritis nach Steinbrocker
|
I ev. gelenknahe Osteoporose
II zusätzlich beginnende Knorpel- und Knochendestruktion III zusätzlich beginnende Subluxation/ Fehlstellungen IV Gelenkzerstörungen und -deformierungen, Luxationen, Ankylosen |
|
|
Diagnosekriterien der rheumatoiden Arthritis (ACR)
|
4 von 7
1. Morgensteifigkeit der Gelenke von mind. 1 h Dauer 2. Arthritis von >= 3 Gelenkbereichen 3. Arthritis der Hand- oder Fingergelenke (PIP oder MCP) 4. Symmetrische Arthritis 5. Rheumaknoten 6. Nachweis von Rheumafaktoren i.S. 7. Typische Röntgenveränderungen Kriterien 1-4 müssen >= 6 Wochen vorliegen. |
|
|
Therapie der rheumatoiden Arthritis
|
Hit hard and early!
-Physikalisch -Radiosynoviorthese -Synovektomie -Rekonstruktive Chirurgie -Reha, Selbsthilfe -Medikamente: 1. Glukokortiokoide, z.B. Prednisolon 20mg 2. Basistherapie mit DMARD (disease modifying antirheumatic drugs) a) Immunsuppressive Therapie: Methotrexat (MTX): Folsäureantagonist mit immunsuppressiver Wirkung, 1x/Woche, Folsäure mindert NW; Azathioprin b) Alkylantien: Cyclophosphamid c) Sulfasalazin d) Hydroxychloroquin e) Biologicals (Anti-TNF-Alpha: Infliximab (Remicade), Adalimumab (Humira), u.w.; Tocilizumab (RoActemra)->Ak gegen IL6-R; Rituximab (MabThera)-> antiCD20, B-Zell-Depletion 3. NSAR: symptomatisch |
|
|
Aulter Morbus Still
|
selten!
Yamaguchi-Kriterien: Majorkriterien: -Intermittierendes Fieber >39°C für mind. 1 Woche -Arthralgien mind. 2 Wochen -typisches flüchtiges feinfleckiges, lachsfarbenes, makulopapulöses Exanthem an Rumpf und prox. Extremitäten -Leukozytose mit Neutrophilie Minorkriterien: -Halsschmerzen -Lymphadenopathie/ Splenomegalie -Leberenzyme erhöht -RF und ANA neg. 5 Kriteiren, davon 2 Majorkr. |
|
|
Seronegative Spondylarthritiden
|
1. Ankylosierende Spondilitis (M. Bechterew)
2. Reaktive Arthritis, Reiter Syndrom 3. Psoriasisarthritis 4. Enteropathische Arthritis 5. Undifferenzierte Spondylarthritis Assoziation mit HLA-B27 Fehlen von Rheumafaktoren ("seronegativ") |
|
|
Morbus Bechterew
|
Ankylosierende Spondylitis
90% HLA-B27 Leitsymptom entzündlicher Rückenschmerz: <40LJ, schleichender Beginn, nächtliche Schmerzen mit Erwachen in der 2. Nachthälfte, Morgensteifigkeit, Besserung bei Bewegung im Laufe des Tages -Sakroiliitis (Mennel-Zeichen) -Spondylitis (Schmerzen im thorakolumbalen Übergangsbereich) -Bewegungseinschränkung der WS (FB-Abstand, Thoraxumfangsdifferenz, Schober, Ott) -evtl. periphere Arthritis, entzündliche Enthesopathien, Uveitis ant., AV-Bl. I, Aortitis, IgA-Nephritis Di.: -Röntgen/MRT (Syndesmophyten, Spondylarthritis, "Bambusform") Th.: NSAR, Kortikoide, Sulfasalazin, TNF-Alpha-Blockade |
|
|
Psoriasis Arthritis
|
Betroffen sind 20% der Psoriasis-Patienten, gehäuft bei Adipositas und Rauchern
5 verschiedene Formen: 1. DIP und PIP-Befall 2. Deformierende mutilierende Polyarthritis 3. Symmetrische Polyarthritis 4. Asymmetrische Oligoarthritis 5. Spondylarthritis mit Sakroiliitis HIV-Infektion als möglicher Trigger, schwere Verläufe Trias: 1. Erythrosquamöse Plaques mit siberweißer Schuppung 2. Nagelveränderungen (Tüpfelnägel, Ölflecken, Onycholyse, Krümelnägel) 3. Arthritis Th: Sulfasalazin, Methotrexat, TNF-Alpha; vorsicht mit Steroiden: verbessern Arthritis, verschlechtern oft Psoriasis Betablocker können Psoriasis verschlechtern! |
|
|
Enteropathische Arthritis
|
-CED (Morbus Crohn, Colitis ulcerosa)
-M. Whipple -"Bypass-Arthritis" nach GI-Anastomosen-Op Abgrenzung zu postenterischer Arthritis: Enteropahtische Arthritis ist aktiv wenn auch der Darm aktiv ist. Postenterische Arthritis kommt vor nach Infektionen mit: Yersinien, Shigellen, Campylobacter, Salmonellen, (M. Whipple?) |
|
|
Epidemiologie des Lupus erythematodes
|
-gehäuft bei schwarzer US-Bevölkerung
-überwiegend Frauen im gebährfähigen Alter, w:m 10:1 -gehäuft bei HLA-DR2 und DR3 |
|
|
Klinik des Lupus erythemtodes
|
-Allgemeinbeschwerden (Fieber, Schwäche, Gewichtsverlust)
-Polyarthritis ohne Erosionen , evtl. aber mit Subluxationen/ Fehlstellungen (Jaccoud-Arthropathie) -Myositis -Hautveränderungen (Schmetterlingserythem, diskoider Lupus, Photosensibilität, oronasale Ulzerationen, vernarbende Alopezie) -Pleuritis, Perikarditis -Koronaritis -Endokarditis Libman-Sacks -Lupusnephritis -neurologische Veränderungen (Vigilanzdefekte, Depressionen, Epilepsie, Apoplex, ED-artiger Verlauf) |
|
|
Labor bei SLE
|
-ANA (antinukleäre Antikörper): 95%
-Anti-dsDNS-AK (AK gegen doppelsträngige DNS): 60-90%, Titer korrelieren mit Krankheitsaktivität -Anti-Ro: 60% -Anti-Sm: 10-20% -Antiphospholipid-AK: 35% -oft Coombs-positive AIHA, Thrombopenie, Leukopenie |
|
|
Verlaufsformen des Lupus erythematodes
|
1. Kutaner Lupus erythematodes (lokalisiert - meist am Kopf oder disseminiert: Druckschmerzhafte Plaques von Hyperkeratosen mit rötlich-entzündetem Rand und zentraler Atrophie)
2. Subakuter kutaner Lupus erythematodes: Allgemeines Krankheitsgefühl, Arthalgien, Myalgien, Hautveränderungen, evtl. Sjögren-Syndrom; Mittelstellung hinsichtlich Prognose und Klinik 3. Systemischer Lupus erythematodes (SLE) |
|
|
Welche Medikamente können einen Lupus erythematodes verursachen?
|
-Dihydralazin (Nepresol)
-Methyldopa -Phenytoin (Phenhydan, Zentropil) -Neuroleptika -Etanercept, Infliximab |
|
|
Biologicals zur Behandlung rheumatoider Erkrankungen
|
1. TNF-alpha-AK
-Infliximab (Remicade) -Adalimumab (Humira) -Etanercept (Enbrel) -Certolizumab pegol (Cimzia) -Golimumab (Simponi) 2. Andere: -Tocilizumab (RoActemra): Ak gegen IL6-Rezeptor ->CRP-Produktion wird unterdrückt -Anakinra (Kineret): IL1-Rezeptor-Ak -Rituximab (MabThera): B-Zell-Depletion -Abatacept (Orencia): Hemmung der T-Zell Ko-Stimulation |
|
|
Biologicals in der Tumortherapie
|
1. Monoklonale AK
-Rituximab (MabThera): CD20 (Lymphome, RA) -Alemtuzumab (Mabcampath): CD52 (CLL) -Iritumomab-Tiuxetan (Zevalin): CD20 (Lymphom) -Bevaccizumab (Avastin): VEGF (Kolorektales Ca, Bronchial-Ca, Mamma-Ca, Nierenzell-Ca) -Cetuximab (Erbitux): EGF-Rezeptor (KRK bei unmut. KRAS-Onkogen) -Panitumumab (Vectibix): EGF-R. (KRK) -Trastuzumab (Herceptin): HER-2 (Mamma-Ca) 2. Tyrosinkinaseinhibitoren -Imatinib (Glivec): bcr/abl, KIT, PDGFR (CML, Ph+ ALL, GIST, MDS/MPD) -Sunitinib (Sutent): VEGFR, EGFR, KIT, PDGFR (GIST, Nierenzell-Ca) -Dasatinib (Sprycel): bcr/alb, KIT (CML, Ph+ ALL) -Sorafenib (Nexavar): VEGFR, PDGFR, KIT (Leberzell-Ca, Nierenzell-Ca) -Erlotinib (Tarceva) und Gefitinib (Iressa): EGFR (Pankreas-Ca, NSCLC) -Lapatinib (Tyverb): EGFR, HER-2 (Mamma-Ca) 3. mTOR Inhib (mammalian target of rapamycin) -Everolismus (Afinitor): mTOR (Nierenzell-Ca, Transplantationsmedizin) -Temsirolismus (Torisel): mTOR (Nierenzell-Ca) |
|
|
Long-QT-Syndrom: Kontraindizierte Medikamente (Auswahl)
|
-Amiodaron, Dobutamin, Epinephrin, Sotalex, Flecainid
-Amitriptylin, Citalopram, Haloperidol, Lithium, Olanzapin (Zyprexa), Risperidon (Risperdal), Sertralin (Zoloft) -Odansetron (Zofran), Sibutramin -Salbutamol, Terbutalin (Bricanyl) -Makrolide, Chinolone, Cotrim -Chloroquin, Mefloquin -Fluconazol u. a. Azole -Tamoxifen |
|
|
Therapie des Lupus erythematodes
|
-Kutaner Lupus: Retinoide, Lichtschutz, Kortisoncreme
-SLE: Lichtschutz, NSAR + Hydroxychloroquin, Kortikoide. Bei schwerem Verlauf: Hochdosierte Prednisolon-Stoßtherapie, Immunsupressiva, bei schweren Organmanifestationen Cyclophosphamid. Frühzeitige Behandlung kardiovaskulärer Risikofaktoren. |
|
|
Lupusnephritis
|
Prognosebestimmende Organmanifestation beim SLE, Auftreten bei >40% der Patienten
WHO-Klassifikation: Kl. I: Minimale mesangiale Lupusnephritis Kl. II: Mesangiale proliferative Lupusnephritis Kl. III: Fokale Lupusnephritis Kl. IV: Diffuse Lupusnephritis Kl. V: Membranöse Lupusnephritis Kl. IV: Fortgeschrittene sklerosierende Lupusnephritis Kl.: -asymptomatische Proteinurie/ Hämaturie (Akanthozyten, Erythrozytenzylinder) -akutes nephritische Syndrom -nephrotisches Syndrom -rasch progrediente Glomerulonephritis -chronische Niereninsuffiienz -renoparenchymatöse Hypertonie Nierenbiopsie! Induktionstherapie bei Kl. III-IV: Steroide + Cyclophosphamid, Erhaltungstherapie mit Azathioprin |
|
|
Pathogenese der progressiven systemischen Sklerose
|
-Systemerkrankung des Bindegewebes mit Kollagenanhäufung und Fibrose von Haut und inneren Organen + obliterierende Angiopathie mit Fibrose und Obliteration kleiner Gefäße (Zwiebelschalenangiopathie mit Intimaproliferation)
-begleitende Neurodegeneration |
|
|
Diagnose und Therapie der progressiven systemischen Sklerose
|
Di.:
-ANA (90%), ACA (Anticentromere AK) 60% bei CREST, Anti-SCL70 (40% bei dSSc) -Kapillarmikrokopie (Nagelfalzkapillaren) -Hautbiopsie Th.: -In der ödematösen Frühphase: Steroide (niedrigdosiert, bei hoher Dosis Gefahr der renalen Krise) -Immunsuppressiva -physikalische Maßnahmen -Prophylaxe von Raynaud-Beschwerden: Kälteschutz, Kalziumantagonisten, ACE-Hemmer -Bei trophischen akralen Störungen Prostglandinanaloga (Illoprost, Sildenafil), Endothelinrezeptoranta-gonisten (Bosentan), Statine -Keine Gabe von Beta-Blockern -Bei therapierefraktärem schwerem Verlauf: Hochdosis-Chemotherapie + autologe Stammzelltransplantation (mesenchymale Stammzellen) |
|
|
Sjögren-Syndrom
|
Chronische Entzündung von Tränen- und Speicheldrüsen
Sicca Syndrom: dry eye, dry mouth (Xerophthalmie, Xerostomie) Karies!, Parotisschwellung, Hornhautulcerationen 1. Primäre Form 2. Sekundäre Form bei -rheumatoider Arthritis -Kollagenosen -Hepatitis B und C -PBC weitere Symptome: -Raynaud-Syndrom -Arthritis -Lymphadenopathie -Neigung zu Allergien und Sprue -selten UIP/ LIP, Nierenbeteiligung, MALT-NHL, periphere Neuropathie, ZNS-Bet., Innenohrschwerhörigkeit |
|
|
Diagnose und Therapie des Sjögren-Syndroms
|
Labor:
hohe BSG, Gammaglobulinvermehrung, RF (50%), SS-B-Ak, SS-A-Ak, Kryoglobuline) -Schirmer-Test: Nachweis einer verminderten Tränensekretion durch Einlegen eines Filterpapierstreifens über das Unterlid -Saxon-Test: Messung der Speichelproduktion durch Abwiegen eines Wattebausches Th.: -Behandlung der Grunderkrankung bei sekundären Formen -Kaugummi, künstlicher Speichel, Augentropfen, viel trinken -Pilocarpin-Derivat Salagen -ggf. Hydroxychloroquin -ggf. Immunsuppressiva bei Organbeteiligung |
|
|
Sharp-Syndrom
|
"mixed connective tissue disease"
Relativ gutartig verlaufendes Krankheitsbild mit einer Überlappungssymptomatik aus SLE, Sklerodermie, Polymositis, rheumatoider Arthritis Immer mit Raynaud-Symptomatik, selten schwere Organbeteiligungen |
|
|
Mikroskopische Polyangiitis (MPA)
|
klinisch ähnlich wie Wegener-Granulomatose, histologisch fehlt die granulomatöse Entzündung
Kl.: -70% Nierenbeteiligung, Prognose-bestimmend -Pulmonale Vaskulitis (diffuse alveoläre Hämorrhagie) -Hautveränderungen (40%): subkutane Knötchen, palpable Purpura |
|
|
Kryoglobulinämische Vaskulitis
|
IgM-IgG-Komplexe (monoklonales IgM reagiert mit polyklonalem IgG)
3 Typen: I Monoklonales Kryoglobulin (M. Waldenström) II Mono- und polyklonale Immunglobuline III Polyklonale Immunglobuline (bei rehumatoiden Erkrankungen) 1. Essentielle Kryoglogulinämie 2. Sekundäre Kryoglobulinämie -HCV -maligne Lymphome -Kollagenosen -Plasmozytom, M. Waldenström Kl.: -Akral betonte, palpable Purpura -Arthralgien -membranöse Glomerulonephritis -Neuropathie |
|
|
Kutane leukozytoklastische Angiitis
|
Isolierte Angiitis der Haut ohne systemische Manifestationen
|
|
|
Morbus Behcet
|
Multisystemerkrankung mit dem Bild einer leukozytoklastischen Vaskulitis; einzige systemische Vaskulitis mit dem Befall von Arterien und Venen
Häufig in der Türkei Kl.: -Orale Aphthen, genitale Aphthen, Papulopusteln, Erythema nodosum - Uveitis (80%) -Arthritis (70%) -Granulome, Ulcera des Magen-Darm-Traktes (30%) -ZNS-Vaskulitis, Sinusvenenthrombose -arterielle und venöse Thrombembolien Pathergie-Test: Pustelbildung nach intrakutanem Stich mit 20G-Nadel in 45°-Winkel Th.: -Steroide, Colchizin, Immunsuppressiva -ggf. Cyclophosphamid, TNF-Blocker -IFNa2a |
|
|
Klassische Panarteriitis nodosa (cPAN)
|
Vaskulitis der mittelgroßen Gefäße
Ät.: Hep. B, unbek. Ursachen Kl.: -Fieber, Gewichtsverlust, Nachtschweiß -Muskel- und Gelenkschmerzen -evtl. Darminfarkte -Herzinfarkt/ Schlaganfall beim jüngeren Patienten -Polyneuropathie, Mononeuritis multiplex Di.: Arteriographie der A. lienalis und des Tr. coeliacus mit Nachweis von Mikroaneurysmen; Biopsie |
|
|
Arteriitis temporalis Horton
|
Granulomatöse Arteriitis, 50% mit Riesenzellen im Versorgungsbereich der A. carotis externa
50% der Patienten haben zusätzlich eine Polymayalgia rheumatica. -Pochende Schläfenkopfschmerzen, Schmerzen beim Kauen -Augenbeteiligung mit Erblindungsgefahr! -A. temporalis verhärtet, druckschmerzhaft, pulslos -evtl. Aortenbogensyndrom Hohe BSG, CK normal Sono: Sanduhrförmige Stenose mit typischem Halo ggf. Biopsie der A. temporalis Th.: Prednisolon 40-60mg/d, bei Amaurosis fugax stationäre Therapie mit hochdosierter i.v.-Kortisongabe; ASS ggf. Cyclophosphamid Gabe von Kortison = diagnostisch! |
|
|
Polymyalgia rheumatica
|
Granulomatöse Arteriitis mittelgroßer und großer Arterien, 50% mit Riesenzellen
20% zusätzlich Arteriitis temporalis Horton Klinik der PMR: -Symmetrische heftige Schmerzen im Schulter- u./o. Beckengürtel, besonders nachts, Druckschmerzhaftigkeit der betroffenen Muskeln -Morgensteifigkeit -Bilaterale Bursitis subdeltoidea bzw. subacromialis -Allgemeinsymptome (Fieber, Gewichtsverlust, Nachtschweiß) Klinik der Arteriitis cranialis: -Pochende (Schläfen-)Kopfschmerzen, Schmerzen beim Kauen, "Kiefer-Claudicatio" -Augenbeteiligung bis 40% mit Erblindungsgefahr (Amaurosis fugax) -A. temporalis verhärtet, druckschmerzhaft, evtl. pulslos -Aortenbogensyndrom (Blutdruckseitendifferenz, Schlaganfall, Myokardinfarkt) Sturzsenkung! (BSG >50mm in der ersten Stunde) promptes Ansprechen auf Kortikoide; 10-20mg/d Prednisolon |
|
|
Fibromyalgie-Syndrom
|
Multilokuläres Schmerzsyndrom mit typischen schmerzhaften Druckpunkten (tender points)
typisch bei Frauen zw. 30. und 60.LJ ACR-Kriterien: 1. Schmerz in mind. 3 Körperregionen über 3 Monate mit mindestens 11 von 18 getesteten tender points 2. Vegetative Symptomatik 3. Funktionelle Beschwerden Symptomatische Therapie, möglichst ohne klassische Analgetika |
|
|
Retroperitoneale Fibrose
|
Morbus Ormond
Inflammatorische Fibrose des retroperitonealen Fettgewebes und Ummauerung der abdominalen Aorta, Iliacalarterien und Ureteren Primär oder sekundär (Medikamente - Methysergid, Malignome, Trauma, Radiatio, Infekte) Kl.: -Abgeschlagenheit, Gewichtsverlust -Bauch-, Flanken- oder Rückenschmerzen -Nephropathie Th.: -Steroide -Chirurgische Therapie |
|
|
Chronisches Müdigkeitssyndrom
|
Hauptkriterium:
Medizinisch nicht erklärte Erschöpfungszustände von > 6 Monaten Dauer, neu aufgetreten, durch Pausen nicht wesentlich verbessert, führen zu deutlicher Verringerung der früheren Aktivität, nicht Folge einer Anstrengung Nebenkriterien: -Gedächtnisstörungen -schmerzhafte Lymphknoten -Muskelschmerzen -Gelenkschmerzen -Kopfschmerzen -nicht erholsamer Schlaf -mehr als 24 Stunden Krankheitsgefühl nach Anstrengungen |
|
|
Arthrosen
|
Langsam progrediente, primär nichtentzündliche degenerative Erkrankung des Gelenkknorpels
Stadien: I Klinisch stumm II Aktivierte Arthrose mit akuten Schmerzen III Manifeste Arthrose mit Dauerschmerzen und Funktionsminderung Frühtrias: Anlauf-, Ermüdungs- und Belastungsschmerz Spättrias: Dauerschmerz, Nachtschmerz, Muskelschmerz Rö: -asymmetrische Gelenkspaltverschmälerung -subchondrale Sklerosierung -Geröllzysten und Osteophyten |
|
|
Welche endokrinologische Erkrankung kann subfebrile Temperaturen/ Fieber verursachen?
|
Hyperthyreose
|
|
|
Def. verschiedener Störungen der Diurese und Miktion
|
Polyurie: >2000ml Harn/d
Oligurie: <500ml Harn/d Anurie: < 100ml Harn/d Pollakisurie: Häufiger Harndrang (z.B. bei Cystitis) Algurie: Schmerzhaftes Wasserlassen Strangurie: mit heftigsten krampfartigen Blasenschmerzen einhergehende Miktion Dysurie: Erschwertes Wasserlassen/ schwacher Harnstrahl bei Blasenenleerungsstörungen |
|
|
Proteinurie
|
1. 30-300mg/d oder 20-200mg/l: Mikroalbuminurie (Frühphasen der diabetischen und hypertensiven Nephropathie)
2. bis 1,5g/d: Kleinmolekulare Proteine -> Tubulopathien; großmolekulare Proteine -> geringe Glomerulopathien 3. 1,5 - 3g/d: Klein- und großmeolekulare Proteine: chronische Glomerulopathien, Transplantatniere, Nephrosklerose 4. >3g/d großmolekulare Proteine: Nephrotisches Syndrom |
|
|
Elektrophoretische Differenzierung von Proteinurien
|
-Albumin: Leitprotein der glomerulären Proteinurie; selektiv-glomeruläre Proteinurie
-Albumin + IgG: unselektiv glomeruläre Proteinurie (schwere glomeruläre Schäden) -Beta-2-Mikroglobulin: Glomeruläre Filtration, tubuläre Rückresorption, erhöhte Werte bei tubulären Läsionen -Bence-Jones-Proteine: Prärenale Proteinurie, Leichtketten bei monoklonaler Gammopathie -Myoglobinurie (prärenal): Muskeltrauma Hämoglobinurie (prärenal): Hämolyse -Tamm-Horsefall-Proteine: postrenal, tubulär sezerniert |
|
|
Nierenschwelle für Glukose
|
160-180mg/dl
|
|
|
Woran erkennt man, dass bei einer Hämaturie eine renale Erkrankung vorliegt?
|
-dysmorphe Erythrozyten (Akanthozyten)
-Erythrozytenzylinder -gleichzeitiges Vorhandensein großmolekularer Proteine |
|
|
Urinsediment
|
1. Hämaturie (Akathozyten: renaler Ursprung)
2. Leukozyturie 3. Epithelien (polygonale Zellen meist renaler Herkunft) 4. Zylinder: -Hyaline Zylinder: Proteinurie, mögl. auch bei Gesunden -Erythrozytenzylinder: pathognomonisch für Glomerulonephritis -Leukozytenzylinder: chronische Pyelonephritis -Epithelzylinder: unspezifisch 5. Harnkristalle (unbedeutend) |
|
|
Welches sind die beiden häufigsten Ursachen für ein terminales Nierenversagen?
|
1. diabetische Nephropathie (35%)
2. Glomerulonephritis (15%) |
|
|
IgA-Nephropathie (M. Berger)
|
Immunkomplexerkrankung mit Ablagerung von IgA im Mesangium und in anderen Anteilen des Glomerulums.
Häufigste Form der idiopathischen GN, vorwiegend jüngere Patienten sekundär bei: Purpura Schoenlein Hennoch, SLE, RA, Leberzirrhose, Sprue, monoklonale Gammopathie, Dematitis herpetiformis, Psoriasis Kl.: 1-3 Tage nach unspezifischen Infekten der oberen Luftwege itnermittierende Makrohämaturie Bei 10% Spontanremission, 25% entwickeln innerhalb von 20 Jahren eine terminale Niereninsuffzienz, nach Nierentransplantation 40% Rezidive Th.: -Prophylaxe von respiratorischen Infekten senkt evtl. Frequenz der Makrohämaturie-Episoden -bei Proteinurie/ Hypertonie: ACE-Hemmer oder AT1-Blocker -bei Proteinurie >1g/24h Kortikoide, evtl. Azathioprin oder Cyclophosphamid |
|
|
Syndrom der dünnen Basalmembran
|
Familiär oder sporadisch
In der Licht- und Immunfluoreszenzmikroskopie normale Niere, im Elektronenmikroskop auffällig dünne Basalmembran Kl.: -Persistierende oder intermittierende Hämaturie mit Exazerbation bei Infekten der oberen Luftwege -gute Prognose, nur ein kleiner Teil der Patienten entwickelt eine art. Hypertonie/ progr. Niereninsuffizienz |
|
|
Alport-Syndrom
|
X-chromosomal dominant vererbter Kollagendefekt
Männer: mikroskopische Hämaturie, Proteinurie, progressive Niereninsuffizienz Frauen: keine Niereninsuffizienz Extrarenale Manifestationen: -Innenohrschwerhörigkeit -Lentikonus und rez. Cornealäsionen |
|
|
Akute postinfektiöse Glomerulonephritis
|
Immunkomplexnephritis, meist nach Infekt mit Beta-hämolysierenden Streptokokken
1-2 Wochen nach Streptokokkeninfekt: Volhard Trias: Mikrohämaturie und Proteinurie, Ödeme, Hypertonie fakultativ: Hirnödem (Kopfschmerzen, epileptische Anfälle), hypertone Krise, Lungenödem, Nierenkapselspannung Im Sediment Erythrozytenzylinder, Akanthozyten, Proteinurie (<3g/d) ASL Titer, ADB-Titer, C3 in der ersten Woche erniedrigt Th.: -Penicillin -Bettruhe, Schonung, salzarme und eiweißarme Diät -Nachuntersuchung über mehrere Jahre! Pro.: -Kinder >90% Ausheilung -Erwachsene 50% Ausheilung |
|
|
Rapid progressive Glomerulonephritis
|
Extrakapilläre proliferierende GN mit diffusen Halbmondbildungen
Typ I (10%): Antibasalmembran-RPGN.: Goodpasture Syndrom mit Lungenbeteiligung Typ II (40%) Immunkomplex-RPGN: oft postinfektiös, SLE, Purpura Schoenlein Hennoch; Ablagerung von Immunkomplexen an der glomerulären Basalmembran ("humps") Typ III (50%) ANCA-assoziierte Vaskulitiden: mPAN, Wegener Granulomatose Klinisch rasch progrediente Niereinsuffizienz Th.: Kortisonstoßtherapie, ggf. Cyclophosphamid, andere Immunsuppressiva; bei Typ I Plasmaaustausch über 2-3 Wochen |
|
|
Pathogenese des neprhotischen Syndroms
|
Proteinurie mit IgG-Verlust ( ->Infektanfälligkeit) und AT-Verlust (Thromboserisiko)
-> Hypalbuminämie -> Flüssigkeitsverschiebungen vom Plasma ins Interstitium -> Aktivierung des RAAS -> Salz- und Wasserretention ->Ödeme |
|
|
Erregerspektrum beim Harnwegsinfekten
|
Akuter unkomplizierter HWI:
-E. coli (70-85%) -Proteus mirabilis (10-15%) Komplizierter HWI mit prädisponierenden Risikofaktoren -E. coli (50%) -Proteus mirabilis (10%) -Klebsiellen (15%) -Enterokokken (10%) -Staphylokokken (10%) -Pseudomonas aeruginosa (5%) |
|
|
Def. prädisponierende Risikofaktoren beim komplizierten HWI
|
-Harnabflusstörungen; VUR
-Instrumentation an den Harnwegen und Harnwegskatheter-assoziierte HWI -Abwehrschwäche -Gravidität -Analgetikaabusus -Diabetes, Gicht, Hyperkalzämie, Hypokaliämie |
|
|
Formen der Harnwegsinfekte
|
A Aysmptomatische Bakteriurie
B Symptomatische HWI: -Aktue Zystitis: Schmerzhaft! Dysurie, Algurie, Polakisurie -Akute Pyelonephritis: Fieber, Dysurie, klopfschmerzhaftes Nierenlager; atypische Bilder mit GI-Beschwerden, unklarem Fieber -Chronische Pyelonephritis: Uncharakteristische Symptome: Kopfschmerzen, Abgeschlagenheit, Brechreiz, Gewichtsabnahme, dumpfe Rückenschmerzen |
|
|
Komplikationen der Pyelonephritis
|
1. Eitrige Nephritis
2. Urosepsis 3. Paranephritischer Abszess 4. Pyonephrose 5. Niereninsuffizienz bei chronischer Pyelonephritis 6. Tubuläre Partialfunktionsstörungen mit Polyurie und Polydipsie, Natriumverlustniere, Kaliumverlustniere 7. Hypertonie |
|
|
Nebenwirkungen von Fluorchinolonen
|
-Störung von ZNS und PNS mit Depressionen, Verwirrung, Halluzinationen
-QT-Verlängerung -Sehnenscheidenent- zündungen und -risse -GI-NW -Blutbildveränderungen KI: Kinder (Knorpelschäden) |
|
|
Tubulo-Interstitielle Nierenkrankheiten (TIN)
|
1. Akute TIN: Nicht-glomeruläre Hämaturie, Proteinurie; Ko.: Nierenversagen
Ät.: -Hantaan-Virus -Parainfektiös, z.B. Streptokokken -Sjögren-Syndrom -Medikamente, z.B. NSAR 2. Chronische TIN Kl.: Chronische Niereninsuffizienz Ät.: -Medikamente (Analgetika) -Chemikalien (Cadmium, Blei, Lithium) -Gicht, Hyperkalzämie -Multiples Myelom, Amyloidose |
|
|
Analgetika-Nephropathie
|
Chronische tubulo-interstitielle Nephritis durch Abusus von Mischanalgetika (ASS, Paracetamol, Coffein) oder Phenacetin
->Blockierung der Synthese des vasodilatatorisch wirksamen Protaglandin E2 -> Durchblutungsstörungen, Papillennekrosen Ko.: -Niereninsuffizienz -Papillennekrosen mit Flankenschmerz, Hämaturie und Fieber -Tubulusschädigung mit herabgesetzten Konzentrationsvermögen -erhöhtes Risiko für Urotheliome und Mammakarzinome |
|
|
Aristolochiasäure-Nephropathien
|
1. Endemische Balkan-Nephropathie (chronisch tubulointerstitielle Nierenerkrankung durch Vergiftung mit Aristolochiasäure-haltigem Mehl durch Kontamination mit Osterluzei-Samen)
2. Chinesische Kräuter-Nephropathie (Kontamination mit Osterluzei-Gewächsen) 3. Melamin-Nephropathie (in krimineller Absicht Babynahrung zugemischt, um einen erhöhten Proteingehalt vorzutäuschen) |
|
|
Myelomniere
|
Cast-Nephropathie
Vorkommen bei 30% aller Patienten mit multiplem Myelom Die nicht-resorbierten Leichtketten gelangen in den distalen Tubulus und in die Sammelrohre, wo die Präzipitation von Leichtketten mit Tamm-Horsfall-Protein zur Bildung von Urinzylindern führt, die schließlich eine Tubulusobstruktion verursachen. |
|
|
Renale tubuläre Partialfunktionsstörungen
|
A Störungen des Aminosäuretransportes (z.B. Cystinurie)
B Renale Glukosurie C Störungen des Wasser- und Elektrolyttransportes -Phosphatdiabetes (Vit-D-resistente Rachitis im Kindesalter) -Abnahme des Konzentrationsvermögens mit Polyurie -Renaler Diabetes insipidus -Natriumverlustniere -Kaliumverlustniere -Renal tubuläre Azidose |
|
|
Bartter-Syndrome
|
autosomal rezessiv vererbte Gruppe renaler Tubulusfunktionsstörungen mit hypokaliämischer Alkalose, Salzverlust und Hypotension
Typen I-V Im Erwachsenenalter manifest: Gitelman-Syndrom Pseudo-Bartter-Syndrom: Laxantienabusus oder Diuretikaabusus |
|
|
Def. und Pathogenese der Kontrastmittelnephropathie
|
Def.: Anstieg des Serum-Kreatinin-Wertes auf > 25% des Ausgangswertes
-Vasokonstriktion mit Verminderung der Nierenperfusion -toxischer Effekt an der Tubuluszelle |
|
|
Akutes Nierenversagen
|
Def.:
Akuter Anstieg von Kreatinin um 0,3mg/dl oder um 50% des Ausgangswertes oder vermindertes Urinvolumen 1. Prärenales ANV (60%): Verminderte Nierenperfusion (Exsikkose, Schock, Sepsis, hepatorenales Snydrom) 2. Intrarenales ANV (35%): Tubulusnekrosen, die zu einer Obstruktion der Tubuli durch sich von der Basalmembran ablösende Epithelien in O2-Mangelsituationen durch herabgesetzte NIerenperfusion (ischämisch, toxisch, septisch, Vaskulitis, RPGN, IgA-N., akute interstitielle Nephritis) 3. Postrenales ANV (5%): Abflussbehinderung |
|
|
Akute interstitielle Nephritis
|
-allergisch (NSAR, Sulfonamide, Penicillin)
-parainfektiös (Hanta, CMV, EBV, Leptospirose) Symptome einer allergischen Reaktion wie Exanthem, Eosinophilie, Fieber |
|
|
Klinische Phasen eines ANV
|
I Initialphase
II Manifestes Nierenversagen -Überwässerung, Linksherzinsuffizienz, Lungenödem, Hirnödem -Hyperkaliämie -metabolische Azidose -Urämie (Gastritis, Ulcera, Enzephalopathie, Perikarditis) -Anämie III Polyurische Phase (Cave Verlust von Wasser, Natrium und Kalium) |
|
|
Laborchemische Unterscheidung zwischen prärenalem und intrarenalem ANV
|
Prärenal: Tubuli sind noch funktionstüchtig, es wird ein konzentrierter Urin mit niedrigem Natriumgehalt ausgeschieden
Intrarenal: Mangelnde Rückresorption von Wasser und Natrium -> verdünnter Urin mit hohem Natriumgehalt |
|
|
Folgen der chronischen Niereninsuffizienz
|
1. Versagen der exkretorischen Funktion: Anstieg der Retentionsparameter, osmotische Diurese mit Nykturie, Polyrurie und Polydipsie (Isosthenurie)
2. Störungen im Wasser-, Elektrolyt- und Säure-Basen-Haushalt: Salz- und Wasserretention -> art. Hyptertonie; Hyperkaliämie; metabolische Azidose 3. Abnahme der inkretorischen Nierenfunktion: Renale Anämie (Erythropoietin), renale Osteopathie (Vit. D) 4. Toxische Organschäden -> Steigerung des kardiovaskulären Risikos (Mediaverkalkung, Intimaverkalkung), urämische Polyneuropathie, Enzephalopathie, Muskelfibrillieren (Myopathie), Perikarderguss, Pleuraerguss, Hautjucken |
|
|
Folgen einer lang anhaltenden metabolischen Azidose
|
1. Ossäre Calciumfreisetzung
2. Gastrointestinale Beschwerden (Übelkeit, Erbrechen, Appetitlosigkeit) 3. Hyperkaliämie 4. Subjektives Empfinden von Dyspnoe 5. Zunahme des Eiweißkatabolismus |
|
|
Isosthenurie
|
Konzentrierungsdefekt der Niere
|
|
|
Klinik und klinische Befunde bei chronischer Niereninsuffizienz
|
Frühsymptome: Polyurie, erhöhter Blutdruck, Ödeme, insbes. Lidödeme
Spätsymptome: -renale Anämie (Müdigkeit, verminderte Leistungsfähigkeit, Blässe der Haut); Cafe-au-lait-Farbe der Haut -urämische Gastroenteropathie (Appetitverlust, Übelkeit) -Kopfschmerzen, Sehstörungen (Enzephalopathie) -Hautjucken (Urämie) -Muskelzuckungen (Myopathie) -Parasthesien (Polyneuropahtie) -renale Osteopathie -urämische Perikarditis, Pleuritis Endstadium -Erbrechen -Luftnot (Fluid lung durch Na und Wasserretention) -Rückgang der Urinmenge -Enzephalopathie -vermehrte Blutungsneigung (Thrombopathie) -urämischer Fötor |
|
|
Konservative Therapie der Niereninsuffizienz
|
1. Vermeidung nephrotoxischer Substanzen (Aminoglykoside, Analgetika, NSAR, KM)
2. RR-Einstellung (ACE-Hemmer, AT1-Blocker) 3. Proteinrestriktion 4. Erhöhung der Flüssigkeitszufuhr auf 2-2,5l/d, ggf. + Schleifendiuretika oder sequ. Nephronblockade 5. Azidoseausgleich (Bicarbonat) 6. NaCl-Zufuhr nach Verlust im Urin 7. Prophylaxe/ Behandlung einer Hyperkaliämie 8. Renal elimierte Medikamente richtig dosieren! 9. Prophylaxe und Therapie der renalen Osteopathie 10. Erythropoietin, Ziel-Hb 11-12g/dl 11. Behandlung kardiovaskulärer Risikofaktoren 12. Therapie eines urämischen Pruritus mit selektiver UV-Phototherapie |
|
|
Organtransplantation
|
Impfungen: Regelimpfungen (Polio, Diphtherie, Tetanus); Hep. B, Influenza, Pneumokokken
Postop. Infektionsprophylaxe: Cotrimoxazol (Pneumocystis jirovecii); ggf. Valganciclovir (bei CMV pos. Spender); jeweils 4 Monate Immunsuppression: Anfangs 3er Kombination: Calcineurin-Inhibitor (Tacrolismus oder Ciclosporin A), Interleukin-2-Rezeptorantagonist (Basiliximab oder Daclizumab), Mycofenolat, ggf. Kortison. Im Verlauf Reduktion auf 2er-Kombination |
|
|
Calcium und Phosphat bei Niereninsuffizienz
|
Hypocalciämie (wird weniger rückresorbiert) und Hyperphospatämie (wird weniger ausgeschieden)
-> sekundärer Hyperparathyroidismus: Knochenabbau und Gefäßverkalkungen Therapieziele: Calcium-Phosphatprodukt sollte <4,5 mmol2/l2 Calcium 2,1-2,37 mmol/l Phosphat 1,13-178 mmol/l PTH 150-300pg/ml -Diätetische Phosphatrestriktion und Phosphatbinder -Vit. D -Cinacalcet (Calcimimetikum ->Erhöhung der Calcium-Sensibilität der Nebenschilddrüse -> PTH und Calcium-Phosphatprodukt sinken) |
|
|
Stadien der chronischen Niereninsuffizienz
|
0 Erhöhtes Risiko für Niereninsuffizienz, GFR>=90
1 Nierenschädigung bei normaler Nierenfunktion, GFR >= 90 2 Nierenschädigung mit milder Niereninsuffizienz GFR 60-89 3 Mittelschwere Niereninsuffizienz, GFR 30-59 4 Schwere Niereninsuffizienz, GFR 15-29 5 Nierenversagen, GFR <15 |
|
|
Kardiorenales Syndrom - Einteilung nach Ronco 2008
|
Typ 1: Akutes kardiorenales Syndrom (akutes Herzversagen löst akutes Nierenversagen aus)
Typ 2: Chronisches kardiorenales Syndrom (chronische Linksherzinsuffizienz verursacht chronische Niereninsuffizienz; periphere Minderperfusion, Mikro- und Makrovaskulopathie) Typ 3: Akutes renokardiales Syndrom (Akutes Nierenversagen verursacht kardiale Dysfunktion; Überwässerung, Elektrolytentgleisungen, urämische Karditis) Typ 4: Chronisches renokardiales Syndrom (chronische Nierenerkrankung verschlechtert kardiale Funktion; akzelerierte Atherosklerose) Typ 5: Sekundäres kardiorenales Syndrom (Systemerkrankungen führen zu parallelen, unabhängigen Schädigungen von Herz und Niere; Sepsis, Autoimmunerkrankungen, Diabetes) |
|
|
Stadien Nierenzellkarzinom
|
T1 Tumor bis 7cm, auf die Niere begrenzt (T1a <4cm)
T2a Tumor 7-10cm T2b Tumor >10cm T3a Invasion in Nierenvene oder perirenale Infiltration T3b Invasion in V. cava unterhalb des Zwerchfells T3c Invasion in V. cava oberhalb des Zwerchfells T4 Durchbruch der Gerota-Faszie N0 Kein LK-Met. N1 Metastase in 1 reg. LK N2 Met. in > 1 LK M1 Fernmetastasen UICC Stadium I T1N0M0 Stadium II T2N0M0 Stadium III T3N0M0 oder T1-3 N1 M0 Stadium IV T4N0, N1M0 oder jedes T N2M0 oder jedes T jedes N M1 |
|
|
Prognose Nierenzellkarzinom (5-Jahres-Überlebensrate)
|
Stadium I und II bis 90%
Stadium III ohne LK-Befall bis 60%, mit LK bis 30% Stadium IV bei Entfernung solitärer Metastasen bis 40%, ansonsten <5% |
|
|
Therapie Nierenzellkarzinom
|
T1a (<4cm): Nierenteilresektion
TI-III Radikale Nephrektomie (Niere mit Gerota-Faszie + ipsilaterle Nebenniere) Bei multiplen Metastasen: Angiogenese-Hemmer (Bevacizumab) Tyrosinkinasehemmer (Sorafenib) Interferon alpha m-TOR-Inhibitoren (Temsirolimus) |
|
|
Bei welcher genetischen Erkrankung kommen gehäuft Nierenzellkarzinome vor?
|
von-Hippel-Lindau-Erkrankung
(hereditäre Multisystemerkrankung, autosomal dominant; ZNS-Hämangioblastome, Angiomatosis retinae) |
|
|
Paraneoplastische Syndrome bei Nierenzellkarzinom
|
-Hyperkalzämie (PTHrP)
-Hypertonie (Renin) -Polyglobulie (Erythropoietin) -Stauffer-Syndrom (Leberfunktionsstörung) |
|
|
Formen der malignen Nierentumoren
|
1. Nierenzellkarzinom (Grawitz-Tumor)
2. Nephroblastom (Wilms-Tumor); dysontogenetischer Tumor aus undifferenzierten Zellen |
|
|
Polyzystische Nierenerkrankungen
|
1. Autosomal dominante polyzystische Nephropathie (ADPKD): Zystennieren Typ III nach Potter, 1:1000; Man. nach 20. LJ, meist bds., gleichzeitig Leberzysten, (100%), Pankreaszysten (10%) und Hirnbasisaneurysmen! (10%)
-Harnwegsinfekte -renale Hypertonie -Niereninsuffizienz 2. Autosomal rezessiv polyzystische Nephropathie (ARPKD): Zystennieren I nach Potter, 1:20000, die meisten Kinder versterben 3. Zystische Nierendysplasie: Typ II nach Potter, uno- oder bilateral 4. Zystische Nephropathien im Rahmen von Fehlbildungssyndromen 5. Juvenile Nephronophthise 6. Markschwammnieren (nicht vererbar, angeborene medulläre Aufweitung der Sammelrohre -Hyperkalziurie, Nephrolithiasis -Nephrokalzinose |
|
|
Urolithiasis
|
Harnsteinarten:
-Kalziumoxalat (75%) -Infektsteine (10%): Struvite (Magnesium-Ammonium-Phosphat) -Uratsteine (5%) -Kalziumphosphat (5%) -seltene, z.B. Zystin Kalziumhaltige Steine sind auf der Rö-Leeraufnahme zu sehen. Vorkommen gehäuft in "Wohlstandszeiten" und trockenen Gegenden Gehäuft bei Harnstau, HWI. Kl.: Kolikartige Schmerzen, Übelkeit, Erbrechen, Hämaturie Th.: -ESWL (extrakorporale Stoßwellenlithotripsie) -PNL (perkutane Nephrolithotomie) -Thiazide senken den Calciumgehalt im Urin -Uratsteine: Uralyt U -> Urin-pH 6,5-7) -Oxalatsteine: Meiden oxalathaltiger Speisen, Ca, MCT-Fette -Infektsteine: Urin ansäueren mit Methionin |
|
|
Wie hoch ist die Perspiratio insensibilis
|
Bei normaler Körper- und Außentemperatur: 1l/24h
Bei Fieber: pro 1°C erhöhter Körpertemperatur 0,5-1l/24h |
|
|
Osmo- und Volumenregulation
|
1. Vermindertes zirkulierendes Blutvolumen: Barozeptoren in den juxtaglomerulären Zellen der Niere
-> Renin-> Angiotensin II -> Aldosteron => Vasokonstriktion und renale Natrium und Wasserretention 2. Erhöhtes zrikulierende Blutvolumen: Volumenrezeptoren im Herz -> ANP, BNP, CNP => Vasodilatation und Hemmung des RAAS => Renale Natrium- und Wasserausscheidung 3. Osmorezeptoren im Hypothalamus: ADH -> Antidiurese, Wasserretention |
|
|
Ätiologie der Hyperhydratation
|
1. Niereninsuffizienz
2. Herzinsuffizienz 3. Hypoproteinämie -Eiweißverlust (nephrot. Syndrom, exsudative Enteropathie) -verminderte Zufuhr (Hungerödem) -verminderte Albuminsynthese (Leberzirrhose) 4. Regulationsstörungen -Hyperaldosteronismus -Therapie mit Gluko- oder Mineralkortikoiden -SIADH 5. TURP (Intravasale Einschwemmung von elektrolytfreier Spülflüssigkeit) |
|
|
Welche Medikamente können Ödeme verursachen?
|
-Kalziumantagonisten
-Minoxidil -NSAR -Glukokortikoide -Östrogene -Antidepressiva -Glitazone |
|
|
Hyperkaliämie
|
1. Übermäßige Zufuhr
2. Verminderte renale Ausscheidung -Akutes Nierenversagen, chronische Niereninsuffizienz -M. Addison 3. Medikamente: ACE-Hemmer, ATII-Blocker, Spironolacton, NSAR, Cyclosporin A, Amilorid, Triamteren, Cotrimoxazol, Pentamidin 4. Verteilungsstörungen -Azidose -diabetisches Koma (Insulinmangel) -Freisetzung von Kalium bei Zellschaden (große Weichteilverletzungen, hämolytische Krise, Zytostase, Gamstorp-Syndrom) Kl.: -evtl. Parästhesien, Muskelzuckungen, Paresen -EKG-Veränderungen bis hin zu Sinuswellen Th.: -Glukose + Insulin -NaBi -Salbutamol -Kationenaustauschharze (Resonium) -Forcierte Diurese mit Schleifendiuretika -Dialyse Cave Pseudohyperkaliämie |
|
|
Hypomagnesiämie
|
1. angeborene Magnesiumverlusterkrankungen
2. Sekundäre Hypomagnesiämie -einseitige Ernährung, Alkoholismus -Malabsorption -vermehrter Bedarf, z.B. Schwangerschaft -Polyurische Störungen, Diuretika -Pankreatitis -Laxantienabusus -Diabetes mellitus, Hyperthyreose Kl.: -Reizbarkeit, Depressionen -Tetanie -Parästhesien -Extrasystolen, erhöhte Digitalisempfindlichkeit -Koronarspasmen -QT-Verlängerung |
|
|
Magnesiumtherapie
|
-Ventrikuläre Arrhythmien durch Digitalis
-Extrasystolie -Torsade-de-pointes-Tachykardie -Eklampsie -vorzeitige Wehentätigkeit |
|
|
Hypokalzämie
|
1. Normales ionisiertes Kalzium: Hypalbuminämie unterschiedlicher Genese
2. Erniedrigtes ionisiertes Kalzium: a) niedriger Magnesiumspiegel: Alkoholismus, Malabsorption, medikamentös (Schleifendiuretika, Gentamycin, Cisplatin) b) normales Magnesium: -Hypoparathyroidismus, Pseudohypparathyroidismus -Vit. D-Mangel -Pankreatitis -renal tubuläre Azidose -chronische Niereninsuffizienz -Therapie mit Antikonvulsiva Kl.: Hypokalzämische Tetanie: Krampfanfälle bei erhaltenem Bewusstsein, Parästhesien, Pfötchenstellung, Stimmritzenkrampf -Chvostek-Zeichen -Trousseau-Zeichen Th.: Bei Tetanie 10ml 10% Calciumglukonat i.v. Vit. D, Calcium DD Hyperventilationstetanie |
|
|
Hyperkalzämie
|
1. Tumorinduziert
a) Osteolyse b) paraneoplastisch (PTHrP) 2. Primärer Hyperparathyroidismus 3. Vit D Intoxikation 4. Immobilisation 5. Sarkoidose Kl.: -Polyurie, Polydipsie -Nephrolithiasis, Nephrokalzinose -Übelkeit, Erbrechen. Obstipation -Psychose, Somnolenz, Koma Hyperkalzämische Krise: Polyurie, Polydipsie, Erbrechen, Exsikkose, Hyperpyrexie, Somnolenz, Koma, Psychose Th.: -Forcierte Diurese mit NaCl und Furosemid -Bisphosphonate -Glukokortikoide bei M .Boeck, Plasmozytom |
|
|
Welche Größen gehen in die Henderson-Hasselbach-Puffergleichung ein?
|
1. pCO2 im Blut
2. Standardbikarbonat 3. pH |
|
|
Regulierung des Säure-Basen-Haushaltes
|
1. Pufferung (v.a. Bicarbonat)
2. Respiratorische Regelung durch Abatmung von CO2 3. Renale Regulation durch Ausscheidung von Wasserstoffionen (dabei Regeneration von Bicarbonat) |
|
|
Azidose
|
1. Metabolisch
a) Additionsazidose (Ketoazidose (Coma diabeticum), Laktatazidose (Schock, Hypoxie, Metformin, Propofol), Intoxikationen) b) Retentionsazidose (Niereninsuffizienz, distal tubuläre Azidose Typ I) c) Substraktionsazidose (enteraler Bicarbonatverlust (Diarrhoe), renaler Bicarbonatverlust (proximale tubuläre Azidose Typ II) Hyperchlorämische Azidose mit normaler Anionenlücke: Substraktionsazidose Normochlorämische Azidose mit vergrößerter Anionenlücke: Additions- und Retentionsazidose Anionenlücke: Na - (Cl + HCO3), Normbereich 12+-4mmol/l Zu wenig Bicarbonat wird durch Chlorid ausgeglichen. Kl: Hyperkaliämie, verminderte Reaktivität der Gefäßmuskulatur auf Katecholamine, Minderdurchblutung der Niere |
|
|
Quincke-Ödem (Angioödem)
|
Akutes Ödem des tieferen Bindegewebes, meist an Lippen, Augenlidern, Zunge und Rachen.
1. Histamin-vermittelt (oft Urtikaria in der Anamnese; Kortikoide, Antihistaminika): -idiopathisch -ASS, ACE-Hemmer -allergisch -physikalisch 2. C1-Esterase-Inhibitor-Mangel (Ödeme auch an Extremitäten und Stamm, abdominale Schmerzattacken; Icatibant (Bradykinin-B2-Rezeptorantagonist), Berlinert (C1-Inhibitor), FFP)) -autosomal dominant vererbt -erworben (maligne Lymphome, Auto-AK, H.p., Yersinien) |
|
|
Hypernatriämie
|
Meist mit Hypovolämie
-Urinosmolalität >800mosm/kg: extrarenaler Wasserverlust, ungenügende Wasserzufuhr -Urinosmolalität <800mosm/kg: renaler Wasserverlust (zentraler oder nephrogener Diabetes insipidus) Kl.: -Muskeleigenreflexe verstärkt -Ruhelosigkeit -evtl. Krampfanfälle |
|
|
Hypokaliämie
|
1. Verlust
a) intestinal (Diarrhoe, Laxantienabusus) b) renal (Nierenerkrankung, Diuretikatherapie, Hyperaldosteronismus, Lakritzabusus, Hyperkortisolismus, Ampho-B) 2. Verteilungsstörung -Alkalose -Insulin Kl.: Je akuter die Hypokaliämie, desto ausgeprägter die Symptome -Adynamie bis zu Paresen -Obstipation, paralytischer Ileus -Abschwächung bis Fehlen der Reflexe -EKG: Extrasystolen, QT-Verlängerung -Nephropathie mit Polyurie und Polydipsie -Metabolische Alkalose Cave Digitalis bei Hypokaliämie! |
|
|
Alkalose
|
1. Metabolisch
-Verlust von saurem Magensaft -Diuretikatherapie mit Hypokaliämie (bei Kaliummangel wird vermehrt H renal ausgeschieden) -Mineralkortikoidexzess -vermehrte Zufuhr von Bicarbonat 2. Respiratorische Alkalose -verstärkte alveoläre Ventilation |
|
|
Welche Elektrolytentgleisung droht bei Gabe von Bicarbonat?
|
Hypokaliämie
|
|
|
Akute Pankreatitis - Ätiologie
|
1. Gallenwegserkrankungen (biliäre Pankreatitis) (60%)
2. Alkoholabusus (30%) 3. Andere (10%): -Medikamente -hereditäre Pankreatitis (Mutation Trypsinogen oder Serinprotease-Inhibitor) -ERCP -Bauchtrauma, Operation -Virsinfektion (AIDS, Hepatitis) -Duodenaldivertikel, penetrierendes Ulcus vent. oder duodeni -schwere Hypertriglyceridämie -Hyperkalzämie -Ascariden in den Gallengängen -Pankreas divisum -nach Pankreastransplantation -idiopathisch |
|
|
Pankreas divisum
|
Häufigste Fehlbildung des Pankreas: Inkomplette Verschmelzung des dorsalen (Ductus santorini) mit dem ventralen (Ductus Wirsungianus) Ausführungsgang. Beide Gänge münden getrennt ins Duodenum ein (Papilla major, Papilla minor). Durch die relative Enge der Papilla minor gehäuft Pankreatitiden.
|
|
|
Pankreas anulare
|
Ringförmige Verwachsung des Pankreas um das Duodenum
-> Duodenalstenose möglich |
|
|
Verlauf der akuten Pankreatitis; Schweregrade
|
1. Phase: Pankreasödem oder- nekrose
2. Phase: Ausheilung 3. Phase: Fakultativ bei nekrotisierender Pankreatitis: Infektion der Nekrosen, Sepsis, Abszess Schweregrade: I Akute interstitielle Pankreatitis (80-85% der Fälle) II Akute nekrotisierende Pankreatitis mit Teilnekrose (15% Letalität) III Akut nekrotisierende Pankreatitis mit Totalnekrose (Letalität >50%) |
|
|
Klink der akuten Pankreatitis
|
Leitsymptom: Oberbauchschmerzen mit Anstieg von Pankreasenzymen, Schmerz oft gürtelförmig
Außerdem: -Übelkeit, Erbrechen, Meteorismus, paralytischer (Sub-)Ileus, -Aszites, Fieber, Hypotonie, Schock, EKG-Veränderungen, Pleuraerguss links, Ikterus Komplikationen: Bakterielle Infektion von Nekrosen, Schock, Verbrauchskoagulopathie, ARDS, akutes Nierenversagen, Arrosion von Gefäßen, Milzvenen- und Pfortaderthrombose, Pankreasabszess, Pankreas-Pseudozysten -Cullen-Zeichen: bläuliche Flecken peroumbilikal -Grey-Turner-Zeichen: bläuliche Flecken im Flankenbereich |
|
|
Akute Pankreatitis - prognostisch ungünstige Laborparameter
|
-Leukozytose > 16.000/yl, hohes CRP
-Serum-Calcium < 2mmol/l -Hkt >50% -LDH > 350U/l -Hyperglykämie -Hypoxie -Kreatininanstieg Die Höhe der Lipase und der Amylase gibt keine Hinweis auf die Schwere und Prognose der Erkrankung, wohl aber die Erniedrigung des Serum-Calciums! Prognostischer Score: z.B. Ranson-Kriterien |
|
|
Wann werden falsch hohe Lipase- und Amylasewerte gemessen?
|
Bei Niereninsuffizienz
|
|
|
Leitsymptome des akuten Abdomens
|
1. Heftige Bauchschmerzen
2. Peritoneale Symptomatik (Abwehrspannung) 3. Störung der Darmperistaltik (Meteorismus, Übelkeit, Erbrechen) 4. Schlechter Allgemeinzustand, Kreislaufstörungen |
|
|
Therapie der akuten Pankreatitis
|
1. Intensivstation
2. Nahrungskarenz bis Schmerzfreiheit, früher enteraler Kostaufbau 3. Ersatz von VOLUMEN, Elektrolyten, Glucose 4. Analgetika: Tramadol, Pethidin 5. Thrombembolieprophylaxe 6. PPI 7. Indikation für Antibiotika: Nekrotisierende oder biliäre Verlaufsform, infizierte Pseudozysten, Abszess (-> Carbapeneme oder Ciprofloxacin, beides jeweils + Metronidazol) |
|
|
Chronische Pankreatitis - Klassifizierung und Ätiologie
|
Klassifikation (Marsaille 1984):
a) mit fokaler Nekrose b) mit segmentaler oder diffuser Fibrose c) kalzifizierend 1. Chronischer Alkoholabusus (80%) 2. Idiopathisch (15%) 3. Andere -Medikamente -Hypertriglyceridämie -Hyperparathyroidismus -hereditär -Autoimmunpankreatitis |
|
|
Klinik der chronischen Pankreatitis
|
1. Leitsymptom: Rezidivierender Schmerz, der nicht kolikartig ist, Dauer Stunden bis Tage
2. Nahrungsintoleranz (Fett): Auslösung von dyspeptischen Beschwerden, Übelkeit, Erbrechen und Schmerz 3. Maldigestion: Gewichtsabnahme, Fettstühle, Meteorismus, Diarrhoe 4. Insulinmangeldiabetes 5. Evtl. rezidivierender Ikterus Komplikationen: -Pankreaspseudozysten -Milz- und Pfortaderthrombose -Stenosen des Pankreasgangsystems mit Ausbildung multipler intraduktaler Konkremente (Pankreatilithiasis), Pankreasgangfisteln -Stenose des distalen Ductus choledochus mit (rezidivierendem) Ikterus; Duodenalstenose -Pankreaskarzinom, insbes. bei der hereditären Pankreatitis |
|
|
Diagnostik bei chronischer Pankreatitis
|
-Bestimmung der Pankreas-Elastase-1 im Stuhl (normal >200yg/g, bei Pankreasinsuffizienz <100yg/g)
-Pankreasverkalkungen (Sono, Röntgen, CT, MRT) -Pankreasgangsteine und Kaliberunregelmäßigkeiten der Pankreasgänge in MRCP, ERCP -Nachweis von Komplikationen |
|
|
Therapie der chronischen Pankreatitis
|
-Therapie der akuten Schübe wie bei akuter Pankreatitis
-Pankreasenzymsubstitution (Lipase) -Therapie der endokrinen Pankreasinsuffizienz, cave Hypoglykämie -Stent-Einlage, ESWL, endokopische Behandlung von Pseudozysten und Abszessen -chirurgische Therapie (Drainage, Teilresektion) |
|
|
Welches sind die beiden häufigsten Stoffwechselerkrankungen der weißen Bevölkerung in Europa und den USA?
|
1. Hämochromatose
2. Mukoviszidose |
|
|
Mukoviszidose
|
autosomal rezessiv, defekte Chloridkanäle (CFTR-Gen: Zystische Fibrose Transmembran-Regulator-Protein) -> Bildung zäher Schleimsekrete
Klinik: 1. Mekoniumileus bei Geburt 2. Atemwege: Chronischer Husten, rezidivierende Bronchialinfekte, Bronchiektasen, obstruktives Emphysem, Pneumothorax, allergische bronchopulmonale Aspergillose 3. Exokrine Pankreasinsuffizienz 4. 10% biliäre Zirrhose der Leber 5. Bei Frauen verminderte Fertilität, bei Männern Infertilität Di: Pilocarpin-Schweißtest (Cl-Gehalt des Schweißes >60mmol/l) Th: Autogene Drainage des zähen Bronchialschleims, Substitution von Pankreasenzymen, parenterale Gabe von EDKA, Ursodesoxycholsäure bei biliärer Zirrhose |
|
|
DD erhöhtes CA 19-9
|
1. Pankreaskarzinom
2. Cholestase 3. Entzündliche Magen-Darm-Erkrankungen |
|
|
Pankreaskarzinom
|
95% duktale Adenokarzinome, frühe lymphogene und hämatogene Metastasierung
Kl.: -keine Frühsymptome! -wie bei chronischer Pankreatitis -Ikterus, Courvoisier-Zeichen: prallelastisch tastbare Gallenblase durch Verschluss des Ducuts choledochus -Thromboseneigung -pathologische Glukosetoleranz Di: MRT (mit MRCP und MR-Angiographie) -Endosono, ERCP, CT -Endokopie des Ductus pacreaticus mit Biopsie Th.: Stad. I und II chirurgisch, ggf. adjuvante Chemotherapie mit Gemcitabine palliativ: ggf. Chemotherapie, Stenteinlage, Schmerztherapie Schlechte Prognose! |
|
|
Insulinom
|
Häufigster endokriner Pankreas-Tumor, meist gutartig, meist klein
Kl: Whipple-Trias: 1. Spontanhypoglykämie durch Nahrungskarenz 2. Autonome und neuroglukopenische Symptome 3. Prompte Besserung nach Glukosezufuhr Di: Fastentest |
|
|
Gastrinom
|
Zollinger-Ellison-Syndrom
Meist im Pankreas lokalisiert, meist maligne, häufig bei Diagnosestellung metastasiert. Kl: -Therapieresistente, rezidivierende, oft atypische Ulcera in Magen, Duodenum oder Jejunum -Diarrhoe, Steathorrhoe, da Magensäsure die Lipase inaktiviert Di: Gastrin basal >1000ng/l sind fast beweisend Sekretintest |
|
|
Vipom
|
Verner-Morrison-Syndrom
Sehr seltener, meist maligner Pankreastumor mit vermehrter Bildung von VIP (vasoaktives Polypeptid) WDHH-Syndrom: Wässrige Diarrhoe Hypokaliämie Hypochlorhydrie VIP wirkt wie Choleratoxin |
|
|
Multiple endokrine Neoplasien
|
autosomal dominante Vererbung
MEN I: Wermer-Syndrom: PHPT, Pankreastumor (Gastrinom oder Insulinom), Hypophysentumore MEN 2a: Sipple-Syndrom: C-Zell-SD-Ca, Phäochromozytom, PHPT MEN 2b: Gorlin-Syndrom: C-Zell-SD-Ca, Phäochromozytom, Schleimhautneurinome, Ganglioneuromatose |
|
|
Zeichen der Leberzirrhose
|
-Spider naevi
-Palmarerythem -Lacklippen, Lackzunge -Weißnägel -Ikterus -Kratzeffekte durch Pruritus -Kollateralgefäße im Bereich der Bauchwand -Aszites -Gynäkomastie -femininer Behaarungstyp -Foetor hepaticus |
|
|
Normale Größe der Leber
|
Abstand Lungen-Leber-Grenze und unterer Leberrand: bis 12 cm
sonograpisch: 14 cm in MCL |
|
|
Vitamin K-abhängige Gerinnungsfaktoren
|
Faktor X, IX, VII, II (=Prothrombin-Komplex, merke 1972)
Protein C und Protein S |
|
|
Laborparameter bei Lebererkrankungen
|
1. Indikatoren einer Leberzellschädigung: GPT (ALT; leberspezifisch), GOT (AST), GLDH
De Ritis Quotient: GOT/GPT <1: leichter Leberzellschaden GOT/GPT >1 schwerer Leberzellschaden 2. Cholestaseparameter: GGT, AP AP: Leber, Knochen, Plazenta) 3. Syntheseleistung der Leber: Cholinesterase, Vit-K-abhängige Gerinnungsfaktoren, Albumin 4. Ammoniak 5. Virusserologie und immunologische Diagnostik 6. Tumormarker (AFP) |
|
|
Einteilung des Ikterus
|
1. Hämolytisch (prähepatisch)
2. Hepatozellulär (Parenchymikterus) 3. Cholestatisch/ Verschlussikterus (-> Ikterus, entfärbte Stühle, bierbrauner Urin, Pruritus) |
|
|
Familiäre Hyperbilirubinämiesyndrome
|
1. Erhöhtes indirektes Bilirubin (unkonjugiertes B.)
a) M. Meulengracht oder Gilbert: autosomal dominant, verminderte UDPGT (Uridin-Diphosphat-Glukoronyltransferase, Konjugationstörung mit verminderter Bilirubinaufnahme in die Leberzelle) b) Crigler-Najjar-Syndrom (Fehlen oder starke Verminderung der UDPGT) 2. Erhöhtes direktes Bilirubin a) Dubin-Johnson-Syndrom (autosomal rezessiv, Konjugation normal, Ausscheidungsstörung; w>m, Manifestation manchmal erst in der Schwangerschaft, KI für orale Kontrazeptiva, zentroazinäres Pigment) b) Rotor-Syndrom (Ausscheidungsstörung, KI für Kontrazeptiva) c) Summerskill-Tygstrup (intermittierend intrahepatischer Verschlussikterus bei Kindern und jungen Erwachsenen, gute Prognose) Bis auf Crigler-Najjar Typ I alle gute Prognose |
|
|
Stoffwechsel Bilirubin
|
Abbauprodukt des Häm, Transport an Albumin gekoppelt zur Leber
Durch DPTGT werden Bilirubin und Glukuronsäure zur wasserlöslichen Form konjugiert -> Ausscheidung über die Gallenwege. Im Darm wird Bilirubin zu Urobilinogen reduziert. |
|
|
Lebererkrankungen in der Schwangerschaft
|
1. Idiopathischer Schwangerschaftsikterus (Prognose für die Mutter gut, für das Kind erhöhte perinatale Mortalität und erhöhte Frühgeburtenrate)
2. Virushepatitis (häufigste Ursache) 3. Ikterus bei Hyperemesis gravidarum (gute Prognose) 4. HELPP-Syndrom (haemolysis, elevated liver enzymes, low platelet count (histologisch Thromben in Pfortaderästen, hämorrhagische Lebernekrosen) 5. Akute Schwangerschaftsfettleber (selten, schlechte Prognose!) |
|
|
Welche Infektionskrankheiten gehen mit einer Begleithepatitis einher?
|
1. Viren:
-EBV, CMV -Gelbfieber, Dengue-Fieber, Marburg-Virus, Ebola-Virus 2. Bakterielle Infektionen: -Brucellose -Q-Fieber -Leptospirose (M. Weil) 3. Parasiten: -Malaria -Amöbiasis -Echinokokkose -Bilharziose |
|
|
Virusbestandteile des Hepatitis-B-Virus
|
HBV-DNA
HBs-AG (Surface-Antigen) HBe-AG (Envelope-AG, sekretorische Form des HBc-AG) HBcAG (Core-Antigen) |
|
|
Verlaufsmöglichkeiten der Hepatitis B
|
1. Asymptomatische Hepatitis (65%) oder akute Hepatitis (30%) mit Ausheilung
HBV-DNA, HBsAg -> im Verlauf verschwindet HBsAg und anti-HBs wird positiv. Dazwischen diagnostische Lücke, in der nur anti-HBc-IgM nachweisbar ist. anti-HBc-IgG zeigt den stattgehabten Kontakt zu HBV an. 2. Viruspersistenz (immunkompetente Erwachsene 5%): a) Asymptomatischer HBsAg-Träger: HBsAg, anti-HBc, anti-HBe pos. (anti-HBs negativ, HBeAg negativ, part. Serokonversion b) Aktive Hep. B: HBeAg positive Form oder HBeAg negative Form |
|
|
Wie unterscheidet sich serologisch eine durchgemachte Hepatitis B von einem Zustand nach Impfung?
|
Durchgemachte Hep. B: anti-HBs und anti-HBc-IgG positiv
Z.n. Impfung: anti-HBs pos., anti-HBc-IgG negativ |
|
|
Definition einer chronischen Hepatitis
|
Hepatitis, die nach 6 Monaten nicht ausgeheilt ist.
|
|
|
Natürlicher Verlauf eins HBsAg-Trägers
|
1. Frühe, hochreplikative Phase:
-hohe Transaminasen + histologische Entzündungszeichen -HBV-DNA, HBeAg als Marker für Infektiösität 2. Späte niedrig replikative Phase: Meist werden nur noch Hüllpartikel produziert (HBsAg). Beim Übergang von hoch- zu niedrig-replikativer Phase passagerer Entzündungsschub, Normalisierung der TA, Verschwinden von HBeAg aus dem Serum, Entwicklung von anti-HBe (-> inaktiver und asymptomatischer HBs-Träger, sog. part. Serokonversion) 3. Ausheilung mit Verlust von HBsAg, Ausbildung von anti-HBs, Verschwinden von HBV-DNA |
|
|
Klinik der chronischen Hepatitis B
|
1. Leberzirrhose (20% der Pat. nach 10 Jahren)
2. HCC (Risiko abhängig von der Viruslast, von Pat. mit Leberzirrhose 3%/J) 3. Extrahepatische Manifestationen: -Panarteriitis nodosa -membranoproliferative GN |
|
|
Therapie der Hepatitis B
|
1. Mit Leberzirrhose: Antivirale Therapie bei jeder nachweisbarenViruslast
2. Ohne Leberzirrhose: Viruslast > 10.000/ml und erhöhte TA a) Alpha-Interferon (beendet bei Pat. mit hoher entzündlicher Aktivität und niedriger DNA in 40% die Virusreplikation) b) antivirale Substanzen: Bei Versagen oder KI gegen Interferon; Pat. mit niedriger Entzündungsaktivität -Lamivudin (40% part. Serokonversion) -Entecavir (bis 70%) -Tenofovir (bis 70%) |
|
|
Hepatitis D
|
Inkomplettes RNA-Virus, das für seine Replikation die Hülle (HBsAg) des HBV benötigt.
1. Superinfektion, dabei oft Umstellung der HBV-Infektion von einer replikativen in eine nicht-replikative Form; gelegentlich fulminante Verläufe, meist chronischer Verlauf mit Übergang in einer Zirrhose 2. Simultaninfektion: Schwere akute Hepatitis, jedoch 95% Ausheilung Alpha-Interferon meist erfolglos, antiretrovirale Substanzen in klin. Studien |
|
|
Genotypen der Hepatitis C
|
In Deutschland:
GT 1 78% GT 2/3 18% GT 4 3% GT 5/6 1% Therapieerfolg bei Genotyp 2/3 höher als bei GT 1 |
|
|
Verlauf einer Hepatitis-C-Infektion
|
85% asymptomatisch
15% symptomatisch 75% chronisch Symptomatische Infektionen können in bis zu 50% der Fälle spontan ausheilen. Asymptomatische Infektionen verlaufen meist chronisch. 20% der Pat. mit chronischer Hep. C entwickeln in 20 Jahren eine Leberzirrhose. Davon 1-4%/J HCC. Fulminante Verläufe 1% |
|
|
Extrahepatische Manifestation einer Hep. C-Infektion
|
-essentielle gemischte Kryoglobulinämie
-membranoproliferative und membranöse GN -Hashimoto-T. -Sjögren-Syndrom -Porphyria cutanea tarda -idiopathische Thrombozytopenie |
|
|
Therapie einer Hep. C
|
Akute Hep. C: PEG-IFN über 24 Wochen -> 95% Ausheilung!
Chronische Hep. C: Kombinierte Therapie mit Interferon und Ribavirin GT 2/3 24 Wochen GT 1,4,5,6 48 Wochen RVR: Rapid virological response - HCV-RNA nach 4 Wochen negativ -> evtl. Therapieverkürzung Wen nach 12 Wochen kein signifikantes Ansprechen -> Abbruch SVR: Sustained virological response - HCV-RNA neg. 6 Monate nach Abschluss der Therapie |
|
|
Hepatitis A
|
RNA-Enterovirus, sehr temperatur- und trockenresistent, meist fäkal-orale Infektion
Infektiösität 2 Wochen vor bis 2 Wochen nach Krankheitsbeginn Ikterischer Verlauf bei Erwachsenen 75%, bei Kindern seltener Fulminante Hepatitis 0,2% Pat. >50J Letalität 3% Keine chronische Hepatitis, keine Virusträger |
|
|
Hepatitis E
|
RNA-Virus, fäkal-orale Übertragung
Heilungsraten 98% (Schwangere nur 80%) Fulminanter Verlauf bis 3% (Schwangere bis 20%) |
|
|
Histologie bei chronischer Hepatitis
|
1. Ätiologie
2. Grad der entzündlichen Aktivität 3. Ausmaß der Fibrose |
|
|
NAFLD
|
Nichtalkoholische Fettlebererkrankung
3 Stadien: 1. Steatosis hepatis 2. Steatohepatitis (NASH) 3. Mikronoduläre Zirrhose Ursache meist metabolisches Syndrom und Diabetes mellitus; parenterale Ernährung Medikamente: Amiodaron, Steroide, Tamoxifen, HAART De Ritis-Quotient bei NASH meist < 1 |
|
|
AFLD
|
Alkoholische Fettlebererkrankung
3 Stadien: 1. Steatosis hepatis 2. alkoholische Steatohepatitis (ASH) 3. Mikronoduläre Zirrhose Tox. Grenze Alkohol Männer 40g Ethanol/d Frauen 20g Ko.: Zieve-Syndrom (alkoholtox. Leberschaden, hämolytische Anämie, Hyperlipidämie) Labor: CDT |
|
|
Reye-Syndrom
|
Kinder bis 15 Jahre nach respiratorischem Infekt und Einnahme von ASS
Diffuse Mitochondrienschädigung mit hoher Letalität, 30% neurologische Schäden |
|
|
Toxische Leberschädigung
|
Man unterscheidet
a) obligat Hepatotoxine (dosisabhängig), z.B. Paracetamol b) fakultative Hepatotoxine (Mehrzahl) Bsp.: 1. Akute/ chronische Hepatitis: INH, Methyldopa 2. Fulminante Hepatitis: Halothan, Paracetamol-Intox. 3. Fettleber: Ehtanol, Tetrazykline 4. Intrahepatische Cholestase: Thyreostatika, Pille 5. Mischtyp aus Cholestase und Hepatitis: Sulfonamide 6. Lebertumoren: FNH und Andenome durch östrogenhaltige Kontrazeptiva |
|
|
Ätiologie Leberzirrhose
|
1. Alkohol
2. Virushepatitis 3. Autoimmunhepatitis 4. PBC, PSC 5. Toxische Lebererkrankungen 6. Stoffwechselerkrankungen: Hämochromatose, M. Wilson, Alpha-1-Antitrypsinmangel, Mukoviszidose 7. Kardiale Zirrhose 8. Budd-Chiari-Syndrom (Verschluss der Lebervenen) 9. Bilharziose |
|
|
Child-Pugh-Kriterien
|
Albumin (g/dl) >3,5 (1), 2,8-3,5 (2), <2,8 (3)
Bilirubin (mg/dl) <2 (1), 2-3 (2), >3 (3) Quick (%) >70 (1), 40-70 (20), <40 (3) Aszites keiner (19), leicht (2), mittelgrad. (3) Enzepahlopathie keine (1), I-II (2), III-IV (3) Child A= 5-6 (1J ÜR 100%) Child B=7-9 (1J ÜL 85%) Child C=10-15 (1J ÜL 35%) |
|
|
Portale Hypertonie - Einteilung nach Ätiologie
|
1. Prähepatischer Block (Thrombose der Pfortader durch Hyperkoaguabilität, Pfortaderkompression)
2. Intrahepatischer Block (>90%) a) präsinusoidal: Bilharziose, myeloproliferative Erkrankungen, Sarkoidose, PBC; Infiltration der Periportalfelder b) sinusoidal: Leberzirrhose c) postsinusoidal: Budd-Chiari-Syndrom 3. Posthepatischer Block: Rechtsherzinsuffizienz, Perikarditis constrictiva |
|
|
Definition portale Hypertonie
|
Druckerhöhung > 12mmHg
|
|
|
Hepatorenales Syndrom
|
Vermindertes HZV, Vasodilatation im Splachnicusgebiet
->RAAS ->renale Vasokonstriktion -> Nierenversagen Typ I: Rasch progredient Typ II: langsam progressiv Auslösende Faktoren: -GI-Blutung -großvolumige Parazentese ohne Ersatz des Plasmavolumens -Forcierte Diuretikatherapie -Spontan bakterielle Peritonitis -Diarrhoe -Nephrotoxische Medikamente Therapie: Terlipressin (2-12mg/d) + Albumin (20-40mg/d) führt zu Vasokonstriktion im Splachnicusgebiet und zur Umverteilung des Blutvolumens -> bessere Nierendurchblutung |
|
|
Klinische Folgen der portalen Hypertonie bei Leberzirrhose
|
1. Ösophagus-, Korpus- und Fundusvarizen
2. Aszites, evtl. SBP 3. Hepatorenales Syndrom 4. Hepatopulmonales Syndrom |
|
|
Pathogenese des Aszites bei Leberzirrhose
|
1. Hypervolämie der Splanchnicusgefäße
2. Hypalbuminämie mit Erniedrigung des kolloidosmotischen Drucks 3. Vermehrte Lymphproduktion 4. Gesteigerte Natriumrückresorption durch RAAS, ADH, Katecholamine |
|
|
Unterscheidung Transsudat - Exsudat beim Aszites
|
Spezifisches Gewicht (Transsudat <1,016g/l)
Eiweißgehalt (Transsudat <2,5g/dl) Serum/Aszites-Albumin-Gradient (Transsudat >1,1g/dl) |
|
|
Ösophagusvarizen
|
Letalität einer Blutung korreliert mit Child-Stadium.
Ohne Rezidivprophylaxe 70% Rezidivblutung innerhalb eines Jahres. Therapie: -Ligatur Methode der Wahl, Sklerosierung hohe Komplikationsrate, Histoacryl bei Magenfundusvarizen -Senkung des portalen Drucks: Somatostatin, Octreotid oder Terlipressin -Infektprophylaxe (Ciprofloxacin) -Prophylaxe Leberkoma durch Lactulose, Eiweißrestriktion -Ultima ratio: Senkstaken-Blakemore-Sonde oder Linton-Nachlasssonde (Fundus) Prophylaxe von Blutungen: a) primär nur bei hohem Blutungsrisiko (Große Varizen, "red colour sign", Child C): Propranolol b) Sekundär: Ligaturen, Propranolol, TIPSS |
|
|
TIPSS
|
Transjugulärer intrahepatischer portosystemischer Stent Shunt: Verbindung zwischen Pfortader und Lebervene -> das im portalvenösen System gestaute Blut kann in den großen Kreislauf abfließen
chirurgisches Verfahren: Warren Shunt |
|
|
Therapie des Aszites bei Leberzirrhose
|
Basistherapie (Stufe 1):
-Bettruhe -Natriumrestriktion -Bei Hyponatriämie Flüssigkeitsrestriktion -Albuminzufuhr, wenn S.-Albumin <3g/dl 2. Stufe (wenn nach 4 Tagen kein Gewichtsverlust) -Spironolacton 100 bis max. 400mg/d -Schleifendiuretikum (Torasemid 10mg bis max. 40mg) -Gewichtsabnahme von max. 500-1000g/d 3. Stufe Parazentese (Indikationen: Gespanntes, schmerzhaftes Abdomen, Atemnot, Natriumexkretion <10mmol/d, Diuretikaresistenz) 4. Stufe TIPSS (bei >3 Parazentesen pro Monat oder Intoleranz des Patienten gegenüber Parazentese) |
|
|
Therapie spontan bakterielle Peritonitis
|
Cephalosporine 3. Gen. oder Gyrasehemmer
|
|
|
Hepatische Enzephalopathie - auslösende Faktoren
|
-vermehrte Ammoniakbildung im Darm nach GI-Blutungen oder eiweißreichem Festessen
-Alkalose -> vermehrte Diffusion von Ammoniak ins Gehirn -verstärkter Eiweißkatabolismus bei fieberhaften Infekten -Therapie mit Sedativa |
|
|
Stadien der hepatischen Enzephalopathie
|
0: asymptomatisch
I: beginnende Schläfrigkeit, Konzentrationsstörungen II: stärkere Schläfrigkeit, Flapping Tremor (Asterixis) III: Pat. schläft fast immer, jedoch erweckbar, Foetor hepaticus IV: Leberausfallkoma, Pat. reagiert nicht mehr auf Schmerzreize EEG-Veränderungen ab St. II |
|
|
MELD-Score
|
Abschätzung der 3-Monats-Letalität bei Leberzirrhose für Transplantation
Kreatinin, Bilirubin, INR |
|
|
Therapie der hepatischen Enzephalopathie
|
-Absetzen von Diuretika und Sedativa
-ausreichende Kalorienzufuhr in Form von Kohlehydraten zur Verhinderung eines Eiweißkatabolismus -Lactulose -Lebertransplantation |
|
|
Transplantatabstoßung bei Leber-TX
|
1. Akut: Periportale Hepatitis, nichteitrige Cholangitis, venöse Endothelitis
2. Chronisch: Nichteitrige progressive Destruktion der kleinen Gallengänge ("vanishing bile duct syndrome") |
|
|
Antidot bei Knollenblätterpilzvergiftung
|
Penicillin G
Silibinin |
|
|
Aktues Leberversagen
|
Ausfall der Leberfunktion bei Patienten, die vorher keine chronische Leberkrankheit hatten.
Ät.: -Virushepatitis (B, D, E insbes. bei Schwangeren) -Paracetamol, Halothan, Ecstasy, Knollenblätterpilz -Akute Schwangerschaftsfettleber, HELLP -Schockleber -Budd-Chiari-Syndrom Kl.: -Hirnödem (häufigste Todesursache) -GI-Blutungen, hämorrhagische Diathese -Hypoglykämie durch verminderte Glukoneogenese Met. Alkalose durch verminderte Harnstoffsynthese und verminderten Bicarbonatverbrauch) -Akutes Nierenversagen |
|
|
Häufige Erreger bei spontan bakterieller Peritonitis
|
E.coli
gram-positive Kokken Klebsiellen |
|
|
Gutartige Lebertumoren
|
1. Leberhämangiom (häufigster benigner Lebertumor; sonographisch echoreicher rundlicher TU, glatt begrenzt, Irisblendenphänomen)
2. Fokal noduläre Hyperplasie (FNH) (Assoziation mit östrogenhaltigen Kontrazeptiva; Radspeichenstruktur) 3. Leberzelladenom (große können Komplikationen verursachen, Entfernung wenn >5cm, Beta-Catenin-mutierte Tumoren erhöhtes Malignitätsrisiko) |
|
|
Hepatozelluläres Karzinom
|
Ätiologie:
-Leberzirrhose -Aflatoxin B1 solitär, multizentrisch, diffus infiltrierend Alpha Fetoprotein (AFP) T1 solitär ohne Gefäßinvasion T2 Solitär mit Gefäßinvasion oder multipel <5cm T3a Multipel >5cm T3b Invasion größerer Äste der V. porta oder der Vv. hepaticae T4 Invasion von Nachbarorganen N0 N1 M0 M1 Th.: -Leberteilresektion bei nicht-zirrhotischer Leber -Leber-Tx (Milan-Kriterien: 1 Herd max. 5cm oder 3 Herde max. 3cm) -lokale ablative Verfahren (Radiofrequenzablation, Äthanolinjektion, Laserinduzierte Thermotherapie, Transarterielle Chemotherapie - TACE) Palliativ: Sorafenib |
|
|
DD Zystische Leberläsionen
|
1. Multiple dysontogenetische Zysten
2. Solitäre Leberzysten 3. Zystische Echinokokkose 4. pyogener Abszess; Amöbenabszess 5. Leberhämatom |
|
|
Cholelithiasis
|
-Cholesterinsteine und gemische Steine (80%) (hoher Anteil Cholesterin, niedriger Anteil Gallensäuren)
-Bilirubin- (Pigment-) Steine Risikofaktoren: 6F: female, fair, fat, forty, fertiel, family Klinik: -Gallenkoliken -unspez. Oberbauchbeschwerden -Murphy-Zeichen: Plötzliches schmerzbedingtes Stoppen der tiefen Inspiration Ko.: -Cholangitis, Cholezystitis (Cahrcot-Trias: Schmerzen im rechten Oberbauch, Ikterus, Fieber) -Steinperforation (Obstruktion des Duodenums - Bouveret-Syndrom, Obstruktion des term. Ileums; klin. Trias: Aerobilie, Dünndarmileus, Steinschatten) -Mirizzi-Syndrom: seltene Form des Verschlussikterus, Gallenblasenhydrops oder Stein im GB-Hals führt zu Kompression des benachbarten Ductus hepaticus) -Porzellangallenblase, Spätkompl. Gallenblasenkarzinom |
|
|
ERCP
PTC |
Endoskoisch-retrograde Cholangiopankreatikographie
Perkutane transhepatische Cholangiographie |
|
|
Indikation zur CHE
|
1. Asymptomatische Gallensteine: Behandlung nur wenn
-begleitender Gallenblasenpolyp >10mm -Stumme Porzellangallenblase 2. Symptomatische Cholelithiasis |
|
|
Gallenblasenkarzinom - Risikofaktoren
|
-Cholelithiasis
-chronische Cholezystitis -Salmonellen-Dauerausscheider -Gallenblasenpolypen > 1cm |
|
|
Klatskin-Tumor
|
Gallengangskarzinom nahe der Hepaticusgabel (4 Typen nach Bismuth: I Ductus h. c. ohne Hepaticusgabel, II Beteiligung der Hepaticusgabel, III bis an Segmentabgänge heran, IV sekundäre Segmentabgänge bds. betroffen)
Risiko erhöht bei: -Choledochuszysten -Choledochussteine -PSC -Parasiten Courvoisier-Zeichen: Schmerzloser Ikterus, tastbar vergrößerte Gallenblase Therapie: Evtl. erweiterte Gallengangsresektion mit Hemihepatektomie und LK-Dissektion in Zentren; endoskopische Stent-Therapie; palliative Chemotherapie |
|
|
Foetor ex ore - Halitosis
|
Foetor ex ore = Mundgeruch
Halitosis = übler Geruch der Atemluft |
|
|
Anatomie des Ösophagus
|
1. Pars cervicalis: Von Ösophagusmund hinter dem Ringknorpel bis Eintritt in die obere Thoraxapertur
2. Pars thoracica: Obere Thoraxapertur bis Zwerchfell 3. Pars abdominalis: Hiatus oesophageus bis Magen 3 physiologische Engen: 1. oberer Ösophagusmund (die am wenigsten dehnbare Stelle) 2. Aortenbogen (hinten) und linker Hauptbronchus (vorne) 3. Hiatus oesophageus Obere 2/3 quer gestreifte Muskulatur, unteres 1/3 glatte Muskulatur |
|
|
Nussknackerösophagus
|
Hyperkontraktiler Ösophagus
Angina-pectoris-artige Beschwerden Dysphagie, Aufstoßen, Sodbrennen Rö.: Perlschnurartig verlaufende Kontraktionen DD Diffuser Ösophagusspasmus |
|
|
GERD -Epidemiologie und Ätiologie
|
Gastroösophageale Refluxerkrankung
NERD: endokopisch negativ (non-reosive reflux diseaese); 60% ERD: erosive reflux disease (40%) 10% der Bevölkerung haben gelegentlich Refluxbeschwerden davon 10% Refluxerkrankung davon 10% Barrett-Ösophagus davon 10% Barrett-Karzinom Ät.: primär oder sekundär (Schwangerschaft, Magenausgangsstenose, Sklerodermie, nach chirurg. Behandlung einer Achalasie) Die meisten Patienten haben eine axiale Hiatushernie. Weitere begünstigende Faktoren: Übergewicht, späte große Mahlzeiten, Alkohol, Kaffee |
|
|
Barrett-Ösophagus
|
Ersatz des Plattenepithels des terminalen Ösophagus durch spezialisiertes Zylinderepithel vom intestinalen Typ mit Becherzellen (Das Zylinderepithel hat eine höhere Proliferationsrate). Der Übergang (Z-Linie) ist unscharf mit Ausziehungen bzw. Inseln von metaplastischem Zylinderepithel in den Ösophagus hinein; 3cm über der gastroösophagealen Grenze
fakultative Präkanzerose, 10% entwickeln Barrett-Karzinom, beim Long-Barett ist das Risiko größer als beim Short-Barrett Long-Barrett >3cm Short-Barett <3cm mikroskopischer Barrett Histologisch Becherzellen LGIN (low-grade IEN) HGIN (high-grade IEN) Chromendoskopie mit Essigsäure oder Methyenblau |
|
|
GERD - Klinik
|
-Sodbrennen
-Luftaufstoßen -Schluckbeschwerden -Regurgitation von Nahrungsresten -Epigastrische Schmerzen -Salziger oder seifiger Geschmack nach Aufstoßen -Zahnschäden -Reizhusten, Refluxbronchitis, Asthma -Heiserkeit (posteriore Laryngitis) Ko.: -Ulzerationen, Blutung -Barrett-Ösophagus |
|
|
Klassifikationen der Refluxösophagitis
|
1. Savary und Miller:
0 Reflux ohne Schleimhautläsionen I Isolierte Läsionen II Longitudinal konfluierende Läsionen III zirkulär konfluierende Läsionen IV Komplikationsstadium 2. MUSE (Metaplasie, Ulkus, Striktur, Erosionen) 3. Los Angeles Klassifikation A Eine oder mehrere Erosionen, die sich nicht zwischen den Kuppen der Mukosafalten erstrecken B Wie A, Erosionen >5mm C Erosionen zwischen 2 oder mehr Kuppen aber <75% der Zirkumferenz D Wie C aber >=75% der Zrikumferenz |
|
|
pH-Metrie bei GERD
|
Pathologischer Befund, wenn Refluxzeiten tagsüber >8%, nachts >3% der Messzeit
|
|
|
Therapie bei GERD
|
1. Allgemeinmaßnahmen:
Gewichtsnormalisierung, kleine fettarme Mahlzeiten, auslösende Noxen meiden (Schokolade, Nikotin, säurehaltioge Getränke, Alkohol, Kaffee, kohlensäurehaltige Getränke, Tomatensoße, Knoblauch), nach dem Essen nicht sofort hinlegen, Schlafen mit hochgestelltem Kopfteil, Verzicht auf die Medikamente, die den Druck im UÖS senken (Anticholinergika, Kalziumantagonisten, Nitro, Theophyllin) 2. PPI 3. H2-Blocker nur bei leichten Beschwerden 4. Fundoplicatio nach Nissen |
|
|
Nebenwirkungen von PPI
|
-Diarrhoe
-Schwindel, Kopfschmerzen, Stimmungsschwankungen -Sehstörungen, Hörstörungen -Veränderungen der Leberenzyme -interstitielle Nephritis -Blutbildveränderungen -Dauertherapie kann zu chronisch atrophischer Gastritis führen -erhöhtes Risiko für Pneumonien |
|
|
Roemheld Syndrom
|
reflektorische Herzbeschwerden durch Meteorismus
|
|
|
Nachkontrollen bei Barrett-Ösophagus
|
Keine IEN: Nach 2 negativen Kontrollen im ersten Jahr alle 3 Jahre bei LSB, alle 4 Jahre bei SSB
Gringgradige IEN: Im Abstand von 6 Monaten, dann jährlich, ggf. endoskopische Mukosaresektion Hochgradigen IEN: Bei sichtbarer Läsion photodynamische Therapie, alternativ: Operation |
|
|
Hiatushernie
|
Kardiofundale Fehlanlage: Stumpfer His-Winkel (ösophagogastraler Winkel)
Axiale Hernie: Kardia oberhalb des Zwerchfells Paraösophagelae Hernie: Ein Teil des Magens schiebt sich mit peritonealem Bruchsack neben die Speiseröhre in den Thorax (Extremvariante: Upside-Down-Magen) Operationsindikation auch im asymptomatischen Stadium |
|
|
Pathophysiologie des gastroösophagealen Reflux
|
1. Insuffiziente Antirefluxbarriere durch den UÖS
- 90% inadäquate Sphincterrelaxationen außerhalb des Schluckaktes, v.a. tagsüber und in aufrechter Körperlage; 10% dauerhaft zu niedriger Druck des UÖS, v.a. nachts im Liegen) -Hiatushernie -Erhöhung des intraabdominalen Drucks 2. Gestörte Selbstreinigung (Clearance) der Speiseröhre (zu wenig Speichel, z.B. bei Rauchern, anticholinerge Wirkung des Nikotins; peristaltische Dysfunktion) 3. Gestörte Mukosaresistenz 4. Aggressives Refluat (z.B. Gallensäuren, Zollinger Ellison Syndrom; evtl. Erstmanifestation nach Eradiaktionstherapie) |
|
|
Ösophagusdivertikel
|
70% Zenker-Divertikel (Pulsionsdivertikel mit Ausstülpung von Mukosa und Submukosa durch eine Lücke in der Muscularis propria oberhalb des oberen Ösophagusspincters (Klian-Dreieck) also eigentlich im Hypopharynx
22% parabronchiale Divertikel 8% epiphrenische Divertikel (oberhalb des unteren Ösophagusspincters; Auftreten im Rahmen von Motilitätsstörungen des unteren Ösophagusspincters) 0,1% der Bevölkerung hat ein Zenker-Divertikel Klinik: Globus-, Druck- oder Fremdkörpergefühl im Hypopharynxbereich, Mundgeruch, Dysphagie, Regurgitation von Speiseresten, Gefahr rezidivierender Aspirationen Th.: Zenker-Divertikel sollte operativ saniert werden |
|
|
Ursachen Ösophagitis
|
1. Infektiös: Candida; HSV, CMV bei AIDS
2. Chemisch: Verätzungen, Reflux von Magensaft, Alkohol 3. Physikalisch: Magensonden, Bestrahlungsfolgen 4. Eosinophile Ösophagitis: atopische Erkrankung |
|
|
Risikofaktoren für die Entwicklung eines Ösophaguskazinoms
|
Adeno-Ca: Barrett-Syndrom
Plattenepithel-Ca: Alkohol, heiße Getränke, Rauchen, Nitrosamine, Aflatoxine Außerdem: -Achalasie, Narbenstenosen nach Lagenverätzungen, Plummer-Vinson-Syndrom bei chronischem Eisenmangel -HPV16 -Z.n. Radiatio |
|
|
Lokalisation und Verteilungshäufigkeit von Ösophguskarzinomen
|
3 physiologische Engen
Oben 15% Mitte 35% Unten 50% |
|
|
TNM- und UICC-Klassifikation des Ösophaguskarzinoms
|
TIS Carcinoma in situ
T1a Lamina propria, Muscularis mucosae T1b Submukosa T2 Muscularis propria T3 Adventitia T4a Pelura, Perikard oder Zwerchfell T4b Aorta, Wirbelkörper oder Trachea N0 Keine LK N1 1-2 LK N2 3-6 LK N3 >=7 LK M0 Ohne Fernmetastasen M1 Mit Fernmetastasen UICC: 0 Tis N0 M0 IA T1; IB T2 IIA T3 N0 M0 IIB T1, T2 N1 M0 IIIA T4a N0 M0, T3 N1 M0, T1, T2 N2 M0 IIIB T3 N2 M0 IIIC jedes T4 jedes N3 M0 IV jedes T jedes N M1 |
|
|
Therapie Ösophaguskarzinom
|
Frühe Adenokarzinome (St. I): Endoskopische Mukosaresektion
St. I und IIA: Radikale Operation mit kurativer Zielsetzung (subtotale Ösophagektomie, komplette Lymphadenektomie, Ösophagusersatz durch Magenhochzug; Operationsletalität 5%) ggf. neoadjuvante Radio- /Chemotherapie ggf. perioperative Chemotherapie bei Barett-Karzinom ggf. Fotodynamische Therapie Palliativ: Metallstents, PEG |
|
|
Welche Zellen produzieren was im Magen?
|
Nebenzellen: Schleim
Hauptzellen: Pepsinogen Beleg- oder Parietalzellen: Säure, Intrinsic Factor |
|
|
Formen der chronischen Gastritis
|
Typ A (5%) = Autoimmungastritis: Im Korpus lokalisiert, Autoimmunerkrankung mit Parietalzell-AK -> Schwund der Parietalzellen (=Belegzellen) führt zu Achlorhydrie, Vit B12-Mangel-Anämie (Perniziöse Anämie)
Typ B (80%)= Bakterielle Gastritis (Helicobacter pylori, Antrumgastritis, aszendierende Ausbreitung, durch Atrophie der Drüsenkörper/ Belegzellen Hypochlorhydrie (nie Achlorhydrie) Typ C (15%) = Chemische Gastritis: NSAR und/ oder Gallereflux Sonderformen: -Crohn-Gastritis -Eosinophile Gastritis -Lymphozytäre Gastritis |
|
|
Histologische Parameter der Gastritis
|
1. chronische Gastritis (Kennzeichen: Infiltration der Lamina propria mit Lymphozyten und Plasmazellen)
2. Aktivität der Entzündung (Dichte der neutrophilen Granulozyten) 3. Atrophie der Drüsenkörper 4. Intestinale Metaplasie 5. H.p.-Besiedlung |
|
|
Komplikationen der H.p.-Gastritis
|
-Entwicklung einer Typ-A-Gastritis
-Ulcus duodeni (5%), Ulcus ventriculi (1%) -Magenkarzinom -Gastrale B-Zell- oder MALT-Lymphome -Idiopathische chronische Urtikaria -Idiopathische thrombozytopenische Purpura |
|
|
Diagnostik von H.p.
|
1. Endoskopisch-bioptisch (HUT - Helicobacter-Urease-Test)
2. 13C-Atemtest (13C-Harnstoff wird von Urease des H.p. gespalten, 13CO2 in Ausatemluft) 3. A.p.-Antigennachweis im Stuhl 4. H.p.-IgG-Nachweis im Serum 5. Kultur 6. Histologie (Versilberung, Giemsa, HE) |
|
|
Therapie von H.p.
|
Absolute Indikationen:
-Ulkuskrankheit -MALT-Lymphome des Magens Relative Indikationen: -H.p.-Gastritis, lymphozytäre Gastritis, Ménétrier-Syndrom -Magenkarzinomprophylaxe bei Risikopatienten -Vor Langzeithterapie mit NSAR bei Risikopatienten -Obere GI-Blutung unter NSAR -ITP -Dyspepsie 1. Französische Trippel-Therapie: PPI, Clarithromycin, Amoxicillin 2. Italienische Trippel-Therapie: PPI, Clarithromycin, Metronidazol |
|
|
Riesenfaltengastritis
|
Ménétrier-Syndrom
Riesenfalten >10mm dick bei Luftinsufflation -Eiweißverlust -epigastrische Schmerzen -Diarhoe, Übelkeit, Erbrechen -Ödeme, Aszites, Pleuraergüsse -Anämie -Infektanfälligkeit (Hypogammaglobulinämie) -Thrombembolie (AT-III-Mangel) Ko.: Magenkarzinom Assoziation mit H.p. |
|
|
Definition Erosion - Ulkus im Magen
|
Erosion: Defekt der Magenmukosa, der die Muscularis mucosae nicht durchdringt
Ulkus: Umschriebener Substanzdefekt, der die Muscularis mucosae durchdringt |
|
|
Ursachen der gasgtroduodenalen Ulkuskrankheit
|
1. H.p., insbes. bei Blutgruppe 0 ("Non-secretors")
2. H.p.-negatives Ulkus: NSAR; selten: Zollinger-Ellison-Syndrom, Hyperparathyroidismus 3. Akutes Stressulkus und -erosionen (Intensivmedizin) |
|
|
Lokalisation von gastroduodenalen Ulcera
|
3 Typen des Ulcus ventriculi (Johnson)
Typ I (60%) Kleine Kurvatur proximal des Angulus Typ II (20%) Kombiniertes Ulcus ventr. distal des Angulus + Ulcus duodeni Typ III (20%) Präpylorisch Atypisch lokalisierte Magenulzera im Korpus/ Fundus und an der großen Kurvatur sind immer karzinomverdächtig Ulcus duodeni: Meist Bulbus, distal gelegene sind verdächtig auf Zollinger-Ellison-Syndrom Typ II |
|
|
Klinik Ulcus ventriculi/ duodeni
|
U. duodeni: Spät-, Nacht- und Nüchternschmerz; Besserung nach Nahrungsaufnahme
U. ventriculi: Sofortschmerz nach Nahrungsaufnahme |
|
|
Komplikationen von Magen-/ Duodenalulcera
|
1. Blutung (20%)
2. Perforation (5%) 3. Penetration, z.B. ins Pankreas 4. Narbige Magenausgangsstenose, "Sanduhrmagen" 5. Pylorusinsuffizienz mit Reflux von Galle, Duodenalsaft 6. Karzinomatöse Entartung eines chronischen Ulcus |
|
|
Welche beiden Erkrankungen müssen bei einem H.p.negativen Ulcus ohne NSAR-Anamnese ausgeschlossen werden?
|
1. Zollinger-Ellison-Syndrom (Gastrin basal und nach Provokation mit Sekretin stark erhöht)
2. Primärer Hyperparathyroidismus |
|
|
Welche chirurgischen Verfahren wurden früher bei Ulcus ventr./ duod. angewendet? Welche Komplikationen traten auf?
|
-2/3 Magenresektion mit Gastroduodenostomie (Billroth I) oder Gastrojejunostomie (Billroth II)
-Selektive proximale Vagotomie Ko.: 1. Frühdumping-Syndrom: 20 Minuten nach dem Essen Bauchschmerzen, evtl. Diarrhoe, Schwäche, Schwindel, Tachykardie (Sturzentleerung des Magenstumpfes mit Überdehnung der abführenden Schlinge -> Vagusreiz; passagere Hypovolämie) 2. Spätdumping (2-3 Stunden nach dem Essen Symptome der Hypoglykämie durch überschießende Insulinproduktion) 3. Beschwerden des zu kleinen Magens 4. Afferent-loop-Syndrom (Stau von Gallesekret) 5. Efferent-loop-Syndrom Postvagotomie-Syndrom (Diarrhoe durch Gallensäuren) Vit-B-12-Mangel Magenstumpfkarzinom |
|
|
Warum begünstige Rauchen die Entstehung von gastroduodenalen Ulcera?
|
Rauchen steigert die nächtliche Säureproduktion und reduziert die Speichelsekretion.
|
|
|
Zollinger Ellison Syndrom
|
1. Exzessive Magensäuresekretion
2. Rezidivierende Ulzera im Magen, Duodenom und oberen Jejunum 3. Nicht-Insulin-produzierender Pankreastumor Neuroendokriner Tumore, Gastrinom, Lokalisation in Pankreas oder Duodenum Weitere Symptome: -Wässrige Diarrhoe (Magensekretion kann 8-10l/d betragen) -Steatorrhoe (Inaktivierung der Lipase) -Erbrechen |
|
|
Systemische Mastozytose
|
Autosomal dominant vererbliche Mastzellproliferation in Haut, GI-Trakt, Knochenmark, LK, Milz und Leber
->vermehrte Histamin-Produktion -Flush, Urticaria pigmentosa, Pruritus -RR-Abfall, Fieber, Schüttelfrost, Schock -Kolikartige Abdominalschmerzen, Übelkeit, Erbrechen, Diarrhoe -Asthma, Kopfschmerzen Typisch: rotbräunliches makulopapulöses Exanthem, Darier-Zeichen (Urtikaria nach Reiben der Haut) Knochenmarkspunktion: Mastzellinfiltrate |
|
|
Forrest-Klassifikation der endoskopischen Blutungsaktivität
|
Ia spritzende Blutung
Ib Sickerblutung IIa keine aktive Blutung, jedoch sichtbares Gefäß ("Gefäßstumpf") IIb keine aktive Blutung, Ulkus jedoch mit Koagel bedeckt IIc keine aktive Blutung, Ulkus jedoch mit Hämatin bedeckt III Ulkus ohne Blutungszeichen |
|
|
Was sind Drüsenkörperzysten/ -polypen?
|
Unter PPI-Therapie vermehrte Gastrinsekretion -> Schwellung der Parietalzellen mit apikaler Protrusion in das Drüsenlumen. Harmloser Befund.
|
|
|
Dieulafoy-Gefäßanomalie
|
Besonders dicke und lange Arterie, die meist im oberen Magendrittel geschlängelt in der Submukosa verläuft. Durch druckbedingte Atrophie der darüber liegenden Gefäßwand Atrophie der Tunica mucosa mit Erosion der exponierten Gefäßwand.
->Schwere Blutungen mit hoher Letalität |
|
|
Präkanzerosen des Magens
|
1. Magenpolypen
2. Chronisch atrophische Autoimmungastritis (Typ A) 3. Chronisch atrophische Typ B-Gastritis (H.p.) -> Karzinom von intestinalen Typ durch Verlust der Säuresekretion intestinale Metaplasie und Dysplasie 4. Magenstumpfkarzinom (15-20 Jahre nach Resektion) 5. M. Ménétrier 6. Hereditäre Karzinomsyndrome (Peutz-Jeghers, FAP, HNPCC) Begünstigende Faktoren: -Rauchen -Hohe Nitratgehalt in der Nahrung (geräucherte und gesalzene Speisen): Bakterielle Umwandlung von Nitrat zu Nitrit + Bildung von karzinogenen Nitrosaminen |
|
|
Klassifikation der Adenomkarzinome am ösophagogastralen Übergang (AEG)
|
AEG I 1-5cm oralwärts der Kardia (eigentliches Barrett-Karzinom)
AEGII Tumor in Höhe der Kardia (eigentliches Kardiakarzinom) AEG III 2-5 cm von Kardia entfernt (subkardiales Karzinom) |
|
|
Laurén - Klassifikation der Magenkarzinome
|
1. Intestinaler Typ: Expansiv (polypös) wachsend und gut begrenzt
2. Diffuser Typ: Infiltrativ wachsend und schlecht begrenzt, Neigung zu frühzeitiger LK-Metastasierung 3. Mischtyp |
|
|
Klinik Magenkarzinom
|
-Meist nur diskrete oder fehlende Oberbauchbeschwerden
-Gewichtsabnahme, Leistungsknick, subfebrile Temperaturen -Widerwille gegen Fleisch -Brechreiz -Virchow-Lymphknoten (links supraklavikulär) -Blutung Metastasierung per continuitatem, lymphogen und hämatogen. Krukenberg-Tumor: Abtropfmetastasen in den Ovarien |
|
|
Postgastrektomie-Syndrom
|
1. Früh-Dumping: Zu schnelle Passage von hyperosmolarem Mageninhalt in die abführende Schlinge mit konsekutiv raschem Flüssigkeitseinstrom in das Darmlumen (-> abdominale Beschwerden) und rascher Umverteilung intravasaler Flüssigkeit in den Darm (kardiovaskulär-vasomotorische Symptomatik)
2. Spät-Dumping: Resorption großer Mengen Kohlehydrate in kurzer Zeit -> überschießende Insulinproduktion mit reaktiver Hypoglykämie 3. Afferent-Loop-Syndrom (Typ II): Abflussbehinderung der zuführenden Schlinge -> Aufstau von Pankreassekret und Galle, ggf. bakterielle Fehlbesiedlung (->postprandiales Völlegefühl, Übelkeit, rechtsseitige Oberbauchschmerzen, Besserung nach Erbrechen von Galle/ Pankreassekret; bei bakterieller Fehlbesiedlung Dekonjugation von Gallensäuren gestörte Fett-Resorption, Vit B12-Mangel 4. Efferent-Loop-Syndrom: Völlegefühl, mechanischer ileus 5. Postvagotomie-Syndrom: Vermehrte Gallenblasenfüllung -> chologene Diarrhoe (Cholestyramin) 6. Anämie (Eisen, Vit. B12, Folsäure-Mangel) 7. Osteopathie |
|
|
Wassermelonenmagen
|
GAVE (Gastric antral vascular ectasia)
Gefäßektasien im Magenantrum radiär zum Pylorus verlaufend. 60% der Pat. haben eine Leberzirrhose, außerdem Assoziation mit Autoimmungastritis, Kollagenosen und Raynaud-Syndrom Kl.: okkulte Blutverluste; Therapie: Argon-Plasmakoagulation |
|
|
Ursachen einer oberen GIB
|
1. Ulcus duodeni/ ventriculi (50%)
2. Erosionen 3. Refluxösophagitis 4. Varizen 5. Mallory-Weiss-Syndrom 6. Magenkarzinom 7. GAVE (Wassermelonenmagen - Angiodysplasien) 8. Hämobilie |
|
|
Ursachen einer mittleren GIB
|
1. Dünndarmtumoren
2. M. Crohn 3. Meckel-Divertikel 4. Angiodysplasien 5. Mesenterialinfarkt |
|
|
Ursache einer unteren GIB
|
1. Hämorrhoiden
2. Proktitis, Colitis ulcerosa 3. Karzinom 4. Divertikulose 5. Polypen 6. Infektiöse Kolitis 7. Angiodysplasien 8. Ischämische Kolitis 9. Endometriose |
|
|
Grading von Ösophagus- /Fundusvarizen
|
1 Nur sichtbar
2 Mit Luft vollständig in die Wand eindrückbar 3 Lumeneinengend 4 Lumenausfüllend Endoskopische Kriterien für erhöhtes Blutungsrisiko: -Große Varize (>5mm) -Red Colour Sign = cherry red spots (rötliche Flecken oder Streifen auf Varize) -Magenfundusvarizen |
|
|
Definition Diarrhoe
|
1. zu häufige Stuhlentleerungen (>3x/d)
2. verminderte Stuhlkonsistenz (>75% Wasser) 3. vermehrte Stuhlmenge (>250g/d) |
|
|
Paradoxe Diarrhoe
|
Stenose (z.B. Karzinom) im mittleren Kolon -> nur bakteriell verflüssigter Stuhl kann die Stenose passieren, es werden kleine Portionen übel riechenden Stuhls abgesetzt
|
|
|
Welche Formen der antibiotika-assoziierten Diarrhoe gibt es?
|
1. Osmotische Diarrhoe: Veränderte Dickdarmflora kann Kohlenhydrate nicht ausreichend resorbieren
2. Sekretorische Diarrhoe: Dihydroxy-Gallensäuren werden durch anaerobe Bakterien nicht zu sekundären Gallensäuren dehydroxyliert -> sekretagoge Wirkung; Fettsäuren 3. Clostridium difficile 4. Segmental-hämorrhagische Kolitis: Hypersensitivitätsreaktion nach Penicillingabe |
|
|
Gallensäureverlustsyndrom
|
Ätiologie
1. M. Crohn, Ileumresektion: Ausfall der Gallensäure-Resorption im Ileum -> Gallensäuren gelangen ins Kolon, werden dort bakteriell dekonjugiert, wirken dann toxisch auf das Kolonepithel. 2. Bakterielle Dekonjugation der Gallensäuren bei Blind-Loop-Syndrom Klinik: -Chologene Diarrhoe (kompensiertes GSVS) -Steathorroe bei Ausfall von >100cm des Ileum (Verlust kann nicht mehr durch vermehrte Neusynthese in der Leber ausgeglichen werden) -Cholesteringallensteine (veränderte Gallenzusammensetzung) -Oxalatnierensteine (Ca wird im Darmlumen statt an Oxalat vermehrt an nicht resorbierbare Fettsäuren gebunden, vermehrte Oxalatresorption, vermehrte renale Oxalatexkretion) -Maldigestionssyndrom Th.: -Cholestyramin bei kompensiertem GSVS -bei dekomp. GSVS Fettrestriktion, MCT-Fette -kausale Behandlung (z.B. Tetrazykline bei Blind-Loop-Snydrom) |
|
|
Einteilung der Diarrhoen nach der Pathogenese
|
1. Osmotische Diarrhoe: Kohlenhydratmalabsorption, Glutenallergie, Laxantien; Kennzeichen: Nach dem Fasten hören die Durchfälle auf.
2. Sekretorische Diarrhoe: Infektiös, Gallensäureverlustsyndrom, Fettsäuren, Vipom, villöses Adenom; Kennzeichen: Diarrhoe hört durch Fasten nicht auf. 3. Entzündliche Diarrhoe = Exsudative Diarrhoe infolge Mukosaschäden; infektiös, CED, Kolonkarzinom, Kennzeichen: Oft Blut, Schleim oder Eiterbeimengungen 4. Motilitätsstörungen: Reizdarmsyndrom, nach Magenresektion oder Vagotomie, hormonelle Ursachen, autonome diabetische Neuropathie |
|
|
Infektiöse Erreger bei chronischer Diarrhoe
|
1. Yersinien
2. Amöben 3. Lamblien bei AIDS-Patienten: CMV, Cryptosporidien, Isospora belli, Mykobakterien |
|
|
Komplikationen einer chronischen Obstipation
|
1. Divertikulose
2. Kolonkarzinom 3. Hämorrhoiden |
|
|
Anorektale Obstipation
|
Beckenbodendysfunktion
Die Patienten leiden unter ständigem Stuhldrang und dem Gefühl, sich trotz heftigen Pressens und weichen Stuhls nie vollständig entleeren zu können. Ursache: Kontraktion des M. sphincter ani externus bei Betätigung der Bauchpresse (Anismus). |
|
|
ROM III-Kriterien zur Diagnose der funktionellen Obstipation
|
1. Starkes Pressen
2. Klumpiger oder harter Stuhlgang 3. Gefühl der unvollständigen Entleerung 4. Gefühl der anorektalen Blockierung 5. Manuelle Unterstützung zur Stuhlentleerung 6. Weniger als 3 Stuhlentleerungen pro Woche 7. Weicher ungeformter Stuhl nur unter Laxantientherapie 8. Ungenügende Kriterien für ein Reizdarmsyndrom |
|
|
Melanosis coli
|
Veränderung der Darmschleimhaut bei chronischem Laxantienabusus
|
|
|
Laxantien
|
-Balaststoffe: Leinsamen, Plantago afra oder Plantago ovata Samen
-Osmotisch wirksame L.: Laktulose, Macrogol -Salinische L.: Magnesiumsulfat, Natriumsulfat -Stimulatorische wirksame L.: Bisacodyl (Dulcolax), Natriumpicosulfat (Laxoberal) -> Nur kurzfristige Gabe!! |
|
|
Borborygmi
|
rumorende Darmgeräusche
|
|
|
Gastrointestinale Gasbeschwerden
|
1. Meteorismus (subjektiv: Blähungsgefühl; objektiv: pathologisch vermehrtes Darmgasvolumen)
2. Luftaufstoßen (Rülpsen, Eruktation) und Luftschlucken (Aerophagie) 3. Flatulenz (>= 24 Flatus pro 24 Stunden) Ursachen: -verschluckte Luft (neurotisch, Stress, gesteigerte Salivation, hastiges Essen und Trinken) -gesteigerte intestinale Gasbildung (nicht resorbierbare Kohlenhydrate, faserreiche Kost (->vermehrte Gasbildung infolge bakterieller Zersetzung) Sondenernährung, Sprue, Laktasemangel, exokrine Pankreasinsuffizienz) -verminderte Gasabsorption durch die Kolonflora (portale Hypertonie, Rechtsherzinsuffizienz, Darmatonie, Antibiotika) -Störung der Motilität (Reizdarmsyndrom, Paresen) Kl.: -Völlegefühl, Enge der Kleider -Borborygmi -splenic or hepatic flexure snydrome -Luftaufstoßen, Flatulenz -Roemheld-Syndrom (funktionelle Herzbeschwerden mit A.p., Herzrhyhtmusstörungen durch Zwerchfellhochstand) |
|
|
Ursachen Malassimilationssyndrom
|
1. Maldigestion
-Z.n. Magenresektion -exokrine Pankreasinsuffizienz -Mangel an konjugierten Gallensäuren (Cholestase oder Gallensäureverlustsyndrom bei Ileumresektion, M. Crohn, Dekonjugation von GS durch bakterielle Fehlbesiedlung bei Blind-Loop-Syndrom) 2. Malabsorption -Dünndarmerkrankungen (Sprue, Whipple, M. Crohn, Laktasemangel, chron. Darminfektionen, Amyloidose, Z.n. Radiatio) -Kurzdarmsyndrom (Z.n. Dündarmresektion) -Störung der enteralen Durchblutung 4. Störung der enteralen Lymphdrainage (Lymphangiektasie) 5. Hormonal aktive Tumoren (Zollinger-Ellison-Syndrom, Verner-Morrison-Syndrom, Karzinoid) |
|
|
Was wird nur im Ileum resorbiert?
|
1. Gallensäuren
2. Vit. B 12 |
|
|
Klinik Malassimilationssyndrom
|
1. Chronische Diarrhoe, oft voluminöse Stühle, evtl. Staetorrhoe
2. Gewichtsverlust 3. Mangelsyndrome -Eiweiß: Abmagerung, Ödeme -KH: Gärungsstühle, Flatulenz, Meteorismus, evtl. niedriger BZ -Fettlösliche Vitamine (A - Nachtblindheit, verminderte Tränensekretion, trockene Haut; D Rachitis oder Osteomalazie; K Blutungsneiung -Vit B12, Folsäure, Eisen: Anämie -Kalium: Schwäche -Kalzium: Tetanie, sek. Hyperparathyroidismus 4. endokrine Störungen (Amenorrhoe) 5. Symptome der Grunderkrankung |
|
|
Endokrine Ursachen für Diarrhoe
|
-Hyperthyreose
-Hypothyreose (Obstipation, bakterielle Übersiedlung) -M. Addison -Diabetes mellitus -Zollinger Ellison Syndrom (Säureinaktivierung von Pankreasenzym) -Karzinoid -Medulläres SD-Ca -VIPom -Phäochromozytom |
|
|
Medikamente, die Durchfall verursachen können
|
-Acarbose
-Magnesium, Kalium -Antibiotika -Metformin -Digitalis -Gallensäuren -Colchizin -Laktulose -NSAR -Theophyllin -Zytostatika |
|
|
Wie ist die Steatorrhoe definiert?
|
Stuhlfett >7g/d
|
|
|
Diagnose exokrine Pankreasinsuffizienz und Gallensäuremangel
|
1. exokrine Pankreasinsuffizienz:
-Pankreas-Elastase-1 (<100yg/d Stuhl) -Sekretin-(Wasser und Bicarbonatproduktion) Pankreozymin-(Enzyme) Test ->Bestimmung von Amylase, Lipase, Trypsin und Chymotrypsin im Duodenalsaft 2. 14C-Glykocholat-Test: Orale Gabe von radioaktiv markierter Gallensäure, normalerweise werden 95% im term. Ileum reabsorbiert, Rest gelangt ins Kolon und wird dort bakteriell dekonjugiert, dabei wird radioaktives CO2 frei, das über die Lunge abgeatmet wird; Gallensäureverlustsyndrom führt zu vermehrter Abatmung von 14CO2 |
|
|
Welche Erkrankungen führen zu Hypoproteinämie?
|
1. Enterales Eiweißverlustsyndrom (Verlust aller Eiweißfraktionen! auch Globuline!)
2. Renal: Nephrotisches Syndrom 3. Hepatisch: Leberzirrhose mit verminderter Syntheseleistung |
|
|
Enterales Eiweißverlustsyndrom
|
Ätiologie:
1. Lymphstauung im Bereich des Darms (Lymphangiektasie, maligne Lymphome, M. Whipple; erhöhter Druck bei konstriktiver Perikarditis, Fontan-Operation) 2. Schleimhauterkrankungen (CED, FAP, M. Ménétrier) Der Eiweißverlust betrifft alle Eiweißfraktionen. ->Hypoproteinämische Ödeme Di.: Alpha1-Antitrypsin im Stuhl Th.: Natriumarme, eiweißreiche Kost, Fettrestriktion, MCT-Fette |
|
|
Pseudoallergische Reaktion (PAR)
|
1. PAR durch Histaminintoleranz (Mangel an Diaminoxidase DAO)
2. PAR durch vasoaktive Amine in Nahrungsmitteln (Histamin: Sauerkraut, Käse, Rotwein, Thunfisch; Serotonin: Bananen, Walnüsse; Tyramin: Käse, Fisch, Wein, Hefe, Bananen, Tomaten, Avocados; Phenylethylamin: Schokolade) 3. PAR durch Lebensmittelzusätze (Tartrazin E102, Benzoesäure E214-219; Sulfit E220-227) 4. PAR durch natürlich vorkommende Stoffe: Sulfite (Bier, Wein), Salicylate (Zitrusfrüchte, Nüsse, Weintrauben) 5. PAR durch Natriumglutamat: Chinarestaurant-Syndrom |
|
|
Morbus Whipple
|
Systemische Infektion mit Tropheryma whippelii (Aktinomycet)
Meist Männer zwischen 30 und 60 Jahren Kl.: 1.Diarrhoe, Steathorrhoe, Abdominalschmerzen, Malabsorption, Gewichtsverlust 2. Extraintestinale Symptome: -Arthritis und Sakroiliitis (oft als Erstsymptom) -Fieber -Polyserositis -Vergrößerung der mesenterialen und retroperitonealen LK -braune Hautpigmentierung -Endokarditis -ZNS-Befall Dünndarmbiopsie: Infiltration mit Makrophagen, die PAS-positive Glykoproteine enthalten Th.: Induktion mit Ceftriaxon Erhaltung: 1 Jahr Cotrimoxazol Unbehandelt innerhalb weniger Monate tödlich. |
|
|
Laktoseintoleranz
|
Vorkommen:
Nordeuropa 2% Deutschland 15% Mittemeerraum 25% schwarze Bevölkerung 80% Asiaten 100% 1. Primär: Abnahme der Laktaseaktivität im Kindes-/Jugendalter 2. Sekundär: Sprue oder andere Dünndarmerkrankungen Laktase hydrolisiert Laktose zu Glukose und Glaktose. Unhydrolysierte Lakotse wird im Kolon bakteriell gespalten in CO2, H2 und kurzkettige FS -> Blähungen, Diarrhoe, abdominelle Schmerzen Di.: H2-Atemtest ggf. Laktose-Toleranztest Fermentierte Milchprodukte werden meist in kleinen Mengen vertragen (Joghurt, Buttermilch, Quark). Butter und gereifter Käse enthalten wenig Laktose. |
|
|
Mit welchem Medikament behandelt man GIST? Welche weitere Erkrankung behandelt man damit?
|
Gastrointestinale Stromatumoren entstehen durch Mutation in Tyrosinkinase-Rezeptoren.
Th.: Imatinib (Tyrosinkinase-Inhibitor) CML |
|
|
Morbus Crohn - Definition, Epidemiologie, Prognose
|
Def.: Diskontinuierlich segmental auftretende Entzündung auch der tiefen Wandschichte des gesamten Gastrointestinaltraktes mit häufigster Lokalisation im terminalen Ileum und proximalen Kolon
50% der Crohn-Patienten haben Mutationen des NOD2-Gens auf Chromosom 16 1/3 Stabile Remission 1/3 steroidabhängig 1/3 steroidrefraktär, Immunsuppressiva notwendig |
|
|
Morbus Crohn - Endoskopie, Histolgie
|
Endoskopie:
-skip lesion (segmentaler Befall) -Aphthen -scharf begrenzte, landkartenartige Ulzera -snail tracks (länglich geformte Ulzera) -Pflastersteinrelief -pin-point-lesion (kleinste hämorrhagische Läsionen) Histologie: -Epitheloidzellgranulome -mehrkernige Riesenzellen -Lymphangiektasie |
|
|
Morbus Crohn - Klinik
|
-Abdominalschmerzen, evtl. ähnlich wie bei Appendizitis
-Diarrhoe, oft ohne Blut -Fisteln und anorektale Abszesse -Malabsorptionssyndrom (Vit B12 Mangel, Gallensäureverlustsyndrom) -Darmstenosen -Amyloidose -NW der Kortikoidtherapie Extraintestinal: -Akrodermatitis enteropathica (Zinkmangeldermatitis), Aphthen, Erythema nodosum, Pyoderma gangränosum -Episkleritis, Uveitis, Iritis, Keratitis -Arthritis, ankylosierende Spondylitis -PSC (seltener als bei Colitis ulcerosa) |
|
|
Morbus Crohn - Therapie
|
1. Kortikoide
-topisch mit Budesonid 3x3mg/d -systemisch mit 30-60mg Prednisolon, stufenweise Reduktion 2. Immunsuppressiva: -Azathioprin, bei Intoleranz Methotrexat 3. Biologicals -TNF-Ak (Infliximab, Adalimumab) Fisteln: Metronidazol |
|
|
CED und Nikotin
|
M. Crohn: Verschlechterung
Colitis ulcerosa: protektiv |
|
|
Colitis ulcerosa: Definition, Prognose
|
Def.: Chronisch entzündliche Dickdarmerkrankung mit kontinuierlicher Ausbreitung und mit Ausbildung von Ulcerationen der oberflächlichen Schleimhautschichten
Prognose: 85% Chronisch rezidivierend 10% Chronisch kontinuierlich 5% Akut fulminant |
|
|
Colitis ulcerosa - Klinik
|
-Blutig schleimige Durchfälle
-Abdominalschmerzen, z.T. krampfartig vor Defäkation (Tenesmen) -Gewichtsverlust -Massive Blutung -Toxisches Megakolon -erhöhtes Risiko für kolorektale Karzinome -Amyloidose Extraintestinale Symptome: -Aphthen, Erythema nodosum, Pyoderma gangränosum (seltener als bei M. Crohn) -Iritis, Uveitis, Episkleritis (seltener als bei M. Crohn) -Arthritis, ankylosierende Spondylitis (seltener als bei M. Crohn) -PSC (häufiger als bei M. Crohn) |
|
|
Diversionskolitis
|
In operativ ausgeschalteten Darmsegmenten auftretende hämorrhagische Kolitis (Ursache: Mangel an kurzkettigen Fettsäuren)
|
|
|
Colitis ulcerosa - Endoskopie, Histologie, Röntgen, Befallsmuster
|
Beginn: Distal im Rektum, Ausbreitung nach proximal ins Kolon
Endoskopie: -Diffuse Rötung -Kontaktvulnerabilität -unscharf begrenzte Ulcerationen -Pseudopolypen Histologie: -Kryptenabszesse Röntgen: -langes glattes Rohr (Fahrradschlauch) |
|
|
Früherkennung kolorektales Karzinom bei Colitis ulcerosa
|
Bei Pankolitis >8Jahren oder gleichzeitiger PSC, bei Linksseitenkolitis >15Jahren: jährlich komplette Koloskopie mit Stufenbiopsien, bei IEN (unabhängig vom Dysplasiegrad) Indikation zur Proktokolektomie
|
|
|
Colitis ulcerosa - Therapie
|
A Remissionsinduktion:
1. Mesalazin (5-ASA) bis 4g/d; bei alleiniger Proktitis als Supp. 2. Kortikoide: 40-60mg Prednisolon bis zur Remission, dann stufenweises Ausschleichen über 4 Wochen; ggf. topisch wirksame Steroide 3. Immunsuppressiva: Azathioprin, ggf. Ciclosporin, Tacrolismus, Infliximab, Adalimumab B Remissionserhaltung: 1. langfristig 5-ASA 1-2g/d oral/ lokal 2. E.coli 3. Azathioprin |
|
|
Stufentherapie der Colitis ulcerosa
|
Geringgradige Aktivität (<4 blutige Durchfälle/d, <37°C, geringes Krankheitsgefühl):
5-ASA Mäßiggradige Aktivität (4-6 blutige Durchfälle/d, bis 38°C, deutliches Krankheitsgefühl): zusätzlich Prednisolon Schwere Krankheitsaktivität (>6 blutige Durchfälle/d, >38°C, schweres Krankheitsgfühl, Puls >100/min): Kortikoide i.v., Ciclosporin i.v., parenterale Ernährung; ggf. Biologicals ggf. Kolektomie, wenn nach 3 Tagen keine Besserung |
|
|
Ileoanale Pouch-Operation
|
Bei elektiver Operationsindikation bei Colitis ulcerosa Methode der Wahl
Nach Entfernung der Rektumschleimhaut wird unter Erhaltung des Sphincterapparates ein Dünndarmbeutel (Pouch) mit der anokutanen Grenze verbunden. Spätkomplikation: Pouchitis (-> Metronidazol) |
|
|
Mikroskopische Kolitis
|
1. Kollagene Kolitis: Verdickung des subendothelial gelegenen Kollagenbandes auf >10ym durch verminderten Kollagenabbau, entzündliches Infiltrat in der Tunica propria
2. Lymphozytäre Kolitis: Intraepitheliale Lymphozytose der Dickdarmmukosa Makroskopisch unauffälliger Befund Kl.: -Wässrige Diarrhoe > 4 Wochen -Gewichtsverlust -Abdominelle Schmerzen -Nächtliche Diarrhoe -Meteorismus Th.: Budesonid 9mg/d für 6-8 Wochen NSAR absetzen, können mikroskopische Kolitis begünstigen Chronisch-rezidivierender Verlauf, oft mit langjährigen symptomfreien Intervallen |
|
|
Reizdarmsyndrom
|
4 Symptomvarianten:
1. Diarrhoe-Typ 2. Obstipationstyp 3. Schmerztyp 4. Blähtyp ROM-III-Kriterien: Abdominelle Beschwerden innerhalb der letzten 6 Monate, mindestens 12 Wochen lang, die nicht aufeinander folgen müssen, mit mindestens 2 der der 3 folgenden Kriterien: 1. Defäkation führt zu Besserung der Beschwerden 2. Änderung der Stuhlfrequenz mit Beginn der Beschwerden 3. Änderung der Stuhlbeschaffenheit mit Beginn der Beschwerden |
|
|
Kolon-Divertikulose
|
Industrieländer: Jendseits des 70. LJ haben 60% der Menschen Kolondivertikel
1. Sigmadivertikel (Pseudodivertikel mit Ausstülpung der Darmschleimhaut durch Gefäßlücken der muskulären Darmwand) 2. Coecumdivertikel (oft angeborene echte Divertikel mit Ausstülpung der gesamten Darmwand, gehäuft in Japan) |
|
|
Kolon-Divertikulitis - Klassifikation und Klinik
|
Klassifikation nach Hansen und Stock
St. 0: Asymptomatische Divertikulose St. I: Akute unkomplizierte Divertikulitis St. II: Akute komplizierte Divertikulitis A Peridivertikulitis (Phlegmone) B Gedeckte Perforation, Abszess, Fistel C Freie Perforation St. III: Chronisch rezidivierende Divertikulitis Kl.: -"Linksappendizitis" -Obstipation/ Diarrhoe -evtl. druckschmerzhafte Walze -Coecumdivertikulitis: "Appendizitis trotz Appendektomie" -Blutung -Fisteln -Perforation -Stenose/ Ileus |
|
|
Diagnose Divertikulitis
|
Sono: Targetzeichen
CT, MRT |
|
|
Therapie Divertikulitis
|
Hochakute Divertikulitis:
-Nahrungskarenz, parenterale Ernährung -Breiband-AB (Metronidazol + Chinolon) Leichte Divertikulitis: -schlackenarme Kost -Breitband-AB Bei schweren Komplikationen notfallmäßige Operation (z.B. Hartmann-Op, zweizeitig). Bei Rezidiven ggf. elektive laparoskopische Sigmaresektion |
|
|
FAP
|
Familiäre adenomatöse Polyposis
1:10.000, 1% aller KRK autosomal-dominant erbliche Mutation des APC-Tumorsuppressorgens Multiple (>100) kolorektale Adenome, obligate Präkanzerose mit hohem Entartungsrisiko ab 15. LJ Vorsorgeuntersuchungen ab 12. LJ, Proktokolektomie vor 20. LJ |
|
|
HNPCC
|
Lynch-Syndrom (Hereditäres, nichtpolypöses Kolonkarzinom-Syndrom)
5% aller KRK, autosomal dominante Vererbung, Mutation verschiedener DNA-Reparaturgene, Mikrosatelliteninstabilität KRK-Risiko 80%, medianes Alter des Auftretens: 45 J. >50% im rechten Kolonabschnitt Erhöhtes Risiko für extrakolische Neoplasien: -Endometrium-Ca (60%) -Ovarial-Ca (10%) -Magen-Ca (10%) -Urothel-Ca (2%) Asterdam-Kriterien: -mindestens 3 Familienangehörige mit HNPCC-assoziierten Karzinomen -einer davon Verwandter ersten Grades der beiden anderen -Erkrankungen in mindestens 2 aufeinander folgenden Generationen -Mindestens ein Patient mit der Diagnose eines Karzinoms vor dem 50. LJ |
|
|
Metastasierung von Rektumkarzinomen
|
1. Lymphogen
Oberes Rektumdrittel (12-16cm): 1 Metastasenstraße (paraaortale LK) Mittleres Rektumdrittel (6-12cm): 2 Metastasenstraßen (zusätzlich Beckenwand) Unteres Rektumdrittel (<6cm): 3 Metastasenstraßen (zusätzlich inguinale LK) 2. Hämatogen: Leber, Lunge (nur beim distalen Rektumkarzinom direkt in die Lunge) |
|
|
Wo liegt bei Rektumkarzinomen die Grenze der kontinenzerhaltenden Chirurgie?
|
5cm ab ano
Rektumkarzinome der oberen 2 Drittel: Sphincter-erhaltende anteriore Rektumresektion mit totaler Mesorektumexzision Rektumkarzinom unteres Drittel: Abdominoperineale Rektumextirpation mit Anlage eines endständigen Anus praeter sigmoidalis |
|
|
Adjuvante Chemotherapie beim Kolorektalen Karzinom
|
UICC III (nodal pos. N1-2): 6 Monate Oxaliplatin + 5FU (FOLFOX) oder Capecitabin + Oxaliplatin (XELOX)
|
|
|
Analkarzinom
|
HPV 16 oder 18
gehäuft bei HIV-Patienten Meist Plattenepithelkarzinome Th.: TIS: Lokale Exzision Lokal begrenztes TU-Stadium: Radiochemotherapie (5FU + Mitomycin) Nur bei größeren Karzinomen radikale abdominoperineale Extirpation mit Anlage eines Anus praeter Bei Fehlen inguinaler LK 5-Jahres-Überlebensrate ca. 80% |
|
|
Spaltung des 2. HT
|
-physiologisch: Aortenton vor Pulmonalton bei tiefer Inspiration
-verstärkte Spaltung bei RSB -atemunabhängige Spaltung bei VSD, Pulmonalstenose -paradoxe Spaltung bei schwerer Aortenstenose, LSB, PM |
|
|
Lautstärke der Herztöne
|
1/6 Nur mit Mühe auskultierbar
2/6 Leise, aber sofort hörbar 3/6 Laut, kein Schwirren 4/6 Geräusch mit Schwirren 5/6 Hörbar, wenn nur der Stethoskoprand die Haut berührt 6/6 Hörbar auf Distanz ohne Stethoskop |
|
|
Abakterielle Endokarditis
|
1. E. rheumatica
2. E. Libmann-Sacks (bei SLE) 3. Löffler-Endokarditis 4. Hedinger-Syndrom (bei Karzinoid) |
|
|
Leitsymptome der bakteriellen Endokarditis
|
1. Fieber
2. Herzgeräusch 3. Bakteriämie 4. Splenomegalie 5. Embolien |
|
|
Was muss man bedenken, wenn man bei einem Pat. mit Endokarditis Streptococcus bovis isoliert?
|
60% haben Kolonpolypen/ karzinome -> Koloskopie!
|
|
|
Kutane Zeichen einer bakteriellen Endokarditis
|
1. Petechien
2. Splinter-Blutungen unter den Nägeln 3. Janeway-Läsionen (Hämorrhagische Maculae und Papeln an Handfläche/ Fußsohle) 4. Osler-Knötchen (linsengroße, schmerzhafte rötliche Knötchen, bes. an Fingern und Zehen) |
|
|
Welche Verlaufsformen der Endokarditis unterscheidet man und welche Keime verursachen diese meist?
|
Akute Sepsis: Staphylokokken
Endokarditis lenta: Streptococcus viridans |
|
|
In welchen Situationen muss eine Endokarditisprophylaxe durchgeführt werden?
|
Pat. ohne manifeste Infektion:
1. Zahnbehandlungen 2. Eingriffe am Respirationstrakt (Adeotomie, Tonsillektomie, starre Bronchoskopie) Eine Endokarditisprophylaxe im Rahmen von Eingriffen am Gastrointestinaltrakt oder Urogenitaltrakt wird nur noch bei bestehenden Infekten empfohlen. Resp.-Trakt, Zahnbeh.: Amoxicillin oder Clindamycin GI-Trakt, Urogen.-Trakt: Ampicillin, Piperacillin oder Vancomycin Haut, muskuloskelettales System: Amoxillin oder Clindamycin Bei MRSA Vancomycin |
|
|
Hauterscheinungen bei akutem rheumatischem Fieber
|
-Rheumatische subkutane Knötchen
-Erythema anulare rheumaticum -Erythema nodosum |
|
|
Therapie bei rheumatischem Fieber
|
1. Sanierung des Streptokokkeninfektes: Penicillin V, bei Allergie Makrolid oder Clindamycin
2. Antiinflammatorisch: -ASS 2g/d -Prednisolon 80mg, stufenweise Dosisreduktion, 4-6 Wochen 3. Tonsillektomie im Intervall 4. Rezidivprophylaxe mit Penicillin über mindestens 10 Jahre, max. 25. LJ |
|
|
Typen von Herzklappenprothesen
|
A Kunstklappen
1. Kugel-Käfig-Prothese: Starr-Edwards; langlebig, aber ungünstige Hämodynamik und hohe Thombogenität 2. Kippscheibenprothese: Björk-Shiley, Medtronic Hall, Omnicarbon; geringerer Gradient als Kugel-Käfig-Prothesen 3. Zweiflügelprothese: St. Jude, Sorin, Carbomedics; aktuell bevorzugter Typ B Biologische Klappen 1. Heterograft 2. Kryokonserviertes Homograft 3. Ross-Prozedur |
|
|
Antikoagulation bei Klappenprothesen
|
1. Biologische Klappen: Antikoagulation mit Marcumar für 3 Monate (Ziel INR 2,5-3,5), dann 100mg ASS
2. Kunsklappen: Lebenslange Antikoagulation Ziel INR: -Zweiflügel-AKE 2-3, bei Risikofaktoren (VHF, red. EF, stattgehabte Embolie, Gerinnungsneigung) 2,5-3,5; nach Thrombembolie bis 4,5 -MKE 2,5-3,5 ggf. zusätzlich ASS |
|
|
Mitralklappenstenose - Pathophysiologie
|
Stenose der MK
-> Behinderung der diastolischen linksventrikulären Füllung -> Vergrößerung des LA und im Verlauf Weiterleitung des erhöhten Drucks auf die Lungenvenen -> Reaktive Konstriktion der pulmonalarteriellen Gefäße, bei Überschreitung der Gegenregulation Lungenödem Über die Phase der passiven pulmonalvenösen Hyptertonie entwickelt sich infolge reaktiver pulmonalarterieller Vasokonstriktion, interstitieller Fibrose sowie Umbau der Lungenarteriolen sekundär eine aktive pulmonalarterielle Hypertonie. ->Rechtsherzhypertrophie ->Dilatation des RV ->Rechtsherzinsuffizienz |
|
|
Graham-Steel-Geräusch
|
relative Pulmonalklappeninsuffizienz
|
|
|
Klinik der Mitralklappenstenose
|
1. Folgen der Drucksteigerung in LA: Vorhofflimmern, ggf. mit Thrombenbildung
2. Folgen der Lungenstauung: -Belastungsdyspnoe -nächtlicher Husten -evtl. Hämoptoe mit Herzfehlerzellen (=hämosiderinhaltige Lungenmakrophagen) 3. Folgen der Rechtsherzinsuffizienz -erhöhter Venendruck mit sichtbarer Venenstauung am Hals und unter der Zunge -Stauungsleber, Stauungsniere (Proteinurie), Ödeme der Beine 4. Folgen des verminderten HZV: -Leistungsminderung -perihpere Zyanose mit rötlich-zyanotischen Wangen (Faszies mitralis) |
|
|
Therapie der Mitralklappenstenose
|
1. Diuretika; ACE-Hemmer und AT-Blocker sind kontraindiziert!
2. Möglichst Erhalt SR, ggf. Frequenzkontrolle VHF mit Digitalis 3. Thrombembolieprophylaxe bei VHF; bei SR ab Grad II 4. Invasiv: Mitralklappenvalvuloplastie (Indikation: Symptomatischer Patient bei MKÖF <1,5cm2; asymptomatischer Patient bei pPA >50mmHg erwägen) 5. Chirurgisch: Komissurotomie oder MK-Ersatz (Indikation: Schwer symptomatischer Patient, MKÖF <1,5cm2; gering symptomatischer oder asymptomatischer Patient MKÖF <1cm2) |
|
|
Röntgen Mitralklappenstenose
|
"Stehende Eiform"
-Vergrößerung LA -Erweiterung A. pulm. -Vergrößerung RV |
|
|
Vorlast - Nachlast
|
Vorlast = enddiastolisches Volumen, also das Volumen was sich am Ende der Diastole in den Ventrikeln befindet
Nachlast= Die Kraft, die angewendet werden muss, um den Druck in der Aorta zu überwinden. |
|
|
Ätioloige Mitralklappenstenose
|
99% der Fälle rheumatisches Fieber
|
|
|
Mitralklappeninsuffizienz -Pathophysiologie
|
Regurgitationsvolumen in LA
-> Lungenstauung -> reaktive pulmonale Hypertonie -> Rechtsherzbelastung -> Rechtsherzinsuffizienz Zur Aufrechterhaltung des HZV: Steigerung des Schlagvolumens -> Volumenbelastung -> Hypertrophie und Dilatation des LV |
|
|
Ätiologie Mitralklappeninsuffizienz
|
-Relative MI: Dilatation des Mitralklappenanulus
-Mitralringverkalkung (ältere Pat.) -Z.n. Valvuloplastie -selten nach bakt. oder rheumatischer Endokarditis -degenerative, myxomatöse Veränderungen der Klappensegel (MK-Prolaps-Syndrom, Marfan-S.) -Elongation oder Ruptur von Chordae tendineae (Myokardinfarkt, idiopathisch) -Dysfunktion eines Papillarmuskels (KHK) |
|
|
Graduierung der MI in der Laevokardiographie
|
I Keine komplette Anfärbung des LA
II Komplette Anfärbung des LA nach mehreren Schlägen, KM-Dichte LA<LV III Komplette und dichte Anfärbung des LA, KM-Dichte LA = LV IV Sofortige Anfärbung (nach 1-2 Schlägen), KM-Dichte LA>LV, KM-Reflux in die Lungenvenen |
|
|
Mitralklappenprolaps-Syndrom
|
Barlow-Syndrom
Überdimensionierte Anteile der Mitralklappensegel wölben sich während der Systole in LA. 3-4% der Bevölkerung, nur 10% symptomatisch. w > m, häufig asthenischer Körperbau Ät.: 1. Idiopathisch: Myxematöse Degeneration im Bereich der MK 2. Sekundär (VSD, KHK, DCM, HCM, Myokarditis, Marfan-S., Ehler-Danlos-S.) Kl.: -Rhythmusstörungen -Synkopen -Dyspnoe, schlechte Belastbarkeit -Angina pectoris -Plötzlicher Herztod (1%) -art. Embolien (Winkel zw. post. MK-Segel und linksartrialer Wand) Auskult.: Klicks (Anspannung elongierter Sehnenfäden) Bei Mitralinsuffizienz cave Sport, engmaschige Kontrollen |
|
|
Aortenklappenstenose - Pathophysiologie
|
Druckbelastung des LV
-> konzentrische Hypertrophie -> diastolische Dysfunktion -> Lungenstauung -> durch Linksherzhypertrophie erhöhter myokardialer O2-Bedarf -> Angina pectoris -> reduziertes HZV , Barorezeptoren führen zu Vasodilatation-> Synkopen durch zerebrale Minderperfusion |
|
|
Aortenklappenstenose - Ätiologie
|
-kalzifizierdende AK-Stenose (früher bei bicuspiden Klappen)
-selten kongenitale AS -selten rheumatische AS |
|
|
Aortenklappenstenose - Klinik
|
-Eingeschränkte Belastbarkeit
-Dyspnoe -Angina-pectoris -Schwindel, Synkopen |
|
|
Aortenklappenstenose - Operationsindikation
|
1. Symptomatischer Patient mit schwere AK-Stenose
2. Asymptomatischer Patient mit schwerer AK-Stenose und -reduzierter LV-Funktion (<50mmHg) -Beschwerden bei Belastung -mittel- bis höhergradig verkalkte AK und rasche hämodynamische Progression (Zunahme des AV-Vmax >0,3 m/s/Jahr) -Abfall RR unter Belastung unter den Ausgangswert -schwere LVH ohne art. Hypertonie |
|
|
Aortenklappeninsuffizienz Ätiologie
|
-bakterielle Endokarditis
-Aortendissektion Typ A -bikuspide Klappe -Dilatation der Aortenwurzel und des Klappenrings durch Atherosklerose -Marfan-Syndrom, Ehler-Danlos-Syndrom |
|
|
Aortenklappeninsuffizienz - Pathophysiologie
|
Diastolischer Rückfluss in LV
-> großes Schlagvolumen, das um das Pendelblutvolumen vermehrt ist -> Volumenbelastung LV mit Dilatation und exzentrischer Hypertrophie -> Abnahme der Ventrikelcompliance mit Zunahme des enddiastolischen Ventrikeldrucks |
|
|
Aortenklappeninsuffizienz - Klinik
|
-Minderung der Leistungsfähigkeit
-Synkopen -Herzrhyhtmusstörungen -Angina pectoris |
|
|
Typische Befunde bei Aortenklappeninsuffizienz
|
-Wasserhammerpuls (celer et altus), RRsys hoch, RR diast. niedrig
-pulssynchrones Dröhnen im Kopf -Corrigan-Zeichen: Sichtbare Pulsation der Carotiden -Quincke-Zeichen: Sichtbarer Kapillarpuls nach leichtem Druck auf einen Fingernagel -de Musset Zeichen: Pulssynchrones Kopfnicken |
|
|
Aortenklappeninsuffizienz - Röntgen
|
Großer, nach links ausladender LV, Elongation der Aorta ascendens, prominenter Aortenknopf
"Schuhform" |
|
|
Operationsindikationen Aortenklappeninsuffizienz
|
1. Symptomatischer Patient ab NYHA II oder Angina pectoris
2. Asymptomatischer Patient -EF <50% - EF >50% aber LVEDD >70mm oder LVESD >50mm |
|
|
Angeborene Herzfehler
|
A Azyanotische Vitien
1. Obstruktionen -Pulmonalstenose -Aortenklappenstenose -Aortenisthmusstenose 2. Primärer Links-Rechts-Shunt -VSD -part. Lungenfehleinmündung -ASD -Atrioventrikulärer Septumdefekt -Aortopulmonales Fenster -persistierender Ductus Botalli B Zyanotische Vitien (Rechts-Links-Shunt) -Fallot´Tetralogie -Pulmonalatresie -Double outlet ventricle -Trikuspidalatresie -Komplette Transposition -Truncus arteriosus -Univentrikuläres Herz -Totale Lungenfehleinmündung |
|
|
Pulmonalstenose
|
Formen: Valvulär (10% der angeb. Herzfehler), subvalvulär, supravalvulär, peripher
I delta p <36mmHg II delta p 36-64 III delta p > 64mmHg Druckbelastung RV -> konzentr. Hypertrophie -> Rechtsherzinsuffizienz 2. HT weit gespalten, kleines HZV Klinik: (Belastungs-)Dysnoe, Ermüdbarkeit, Schwindel, Synkopen, Vorhofflattern, Angina pectoris Th.: -Ballonvalvuloplastie/ Stentimplantation -Melody-Klappe -chirurgisch Indikationen: Symptomat. Pat., hoher Gradient, assoziierter ASD oder VSD |
|
|
Aortenisthmusstenose
|
Organische Stenose an der physiologischen Enge zwischen Abgang der A. subclavia sinistra und der aortalen Mündung des Ductus Botalli
-85% mit bikuspider Aortenklappe assoziiert -VSD, ASD -intrakranielle Aneurysmen Perfusion der unteren Körperhälfte über Kollateralgefäße; hoher RR an der oberen Körperhälfte, niedriger RR an der unteren Körperhälfte Letalität infolge Hypertonie und kardiovaskulärer Komplikationen höher als in der Normalbevölkerung. Aneurysmen der Aorta ascendens und/ oder descendens |
|
|
Atrioventrikulärer Septumdefekt (ASVD)
|
veraltet: AV-Kanal
Partieller ASVD: ASD I, Spaltbildung in der Mitralklappe, bindegewebige Verbindung der beiden AV-Klappen Kompletter ASVD: ASD I + VSD, alle 4 Herzhöhlen stehen miteinander in Verbindung |
|
|
Persistierender Ductus arteriosus (Botalli)
|
Verbindung zwischen Pulmonalarterie und Aorta bleibt länger als 3 Monate unverschlossen.
Gehäuft bei Frühgeburten und Rötelnembryopathie Links-Rechts-Shunt -> Volumenbelastung der Lungengefäße, LA, LV. bei großem PDA nahezu keine Drucktrennung mehr. Eisenmenger-Reaktion. Bei kleinem PDA Gefahr der Endarteriitis mit septischen Embolien und Lungenabszessen. Bei mittelgroßen PDA Beschwerden meist erst ab 30. LJ, bei großdem PDA Herzinsuffizienz schon im Säuglingsalter. |
|
|
Formen der Zyanose
|
1. Zentrale Zyanose:
Verminderte O2-Sättigung des arteriellen Blutes ->Haut, Zunge und Mundschleimhaut zyanotisch ->Lewis Test: Nach Massage des Ohrläppchens bleibt es zyanotisch gefärbt -pulmonal bedingt (ungenügende Oxygenierung) -kardial bedingt (Rechts-Links-Shunt) 2. Periphere Zyanose: Vermehrte O2-Ausschöpfung des Blutes in der Kapillarperipherie durch verminderten Blutfluss und Vasokonstriktion (Schock, Herzinsuffizienz, Kälteexposition, Durchblutungsstörungen) ->Zyanose der Akren, nicht der Zunge/ Mundschleimhaut Hämoglobinzyanose unter einem Hb von 5g/dl nicht sichbar! Chronische Hypoxie -> Polyglobulie, hypertrophe Osteoarthropathie (Pierre-Marie-Bamberger) mit Trommelschlegelfingern und Uhrglasnägeln |
|
|
Methämoglobinämie
|
Physiologischer Met-Hb-Gehalt des Blutes <1,5%.
Sichtbare Zyanose ab 10%, Symptome ab 35%. Ursachen: -Glukose-6-Phosphat-Dehydrogenase-Mangel -Intoxikation mit Sulfonamiden, Phenacetin -Gewerbliche Gifte (Nitro- und Aminoverbindungen) Heinz-Innenkörperchen in Erythrozyten, spektroskopische Met-Hb-Bestimmung; Schnelltest: braune Farbe des Blutes |
|
|
Ebstein-Anomalie
|
Fehlbildung eines oder mehrerer TK-Segel mit Apikalverlagerung -> Unterteilung des rechten Herzens in rechten Vorhof, artrialisierten rechten Ventrikel und Restventrikel.
Häufig mit PFO oder ASD assoziiert, häufig WPW-Syndrom, häufig TI. -> Volumenbelastung des RA -> über interartrikuläre Verbindung häufig rechts-links-Shunt |
|
|
Fallot-Tetralogie
|
Verlagerung des Infundibulumseptums nach rechts antero-cephal
-> Obstruktion des rechtsventrikulären Ausflusstraktes -> VSD -> über dem VSD reitende Aorta -> konsekutive Rechtsherzhypertrophie Häufigster zyanotischer angeborener Herzfehler, 10% aller Herzfehler Druckausgleich zwischen RV, LV und Aorta durch den VSD. Wegen RVOTO fließt weniger Blut in die Lunge und gelangt über VSD direkt in den Systemkreislauf. Kl.: -abhängig vom Ausmaß der RVOTO -Belastungsdyspnoe -Hockstellung (-> Anstieg des Systemwiderstandes, Erhöhung der Lungenperfusion) -Hypoxische Anfälle (Engstellung des hypertrophierten Infundibulums) Nach Korrektur Gefahr plötzlicher Herztod durch maligne ventrikuläre Herzrhyhtmusstörungen |
|
|
Transposition der großen Arterien
|
Aorta entspringt aus dem RV, Pulmonalarterie aus dem LV. Überleben nur möglich durch Shuntverbindung (ASD, VSD)
|
|
|
Eisenmenger-Reaktion
|
Pulmonale Hypertonie durch erhöhten Gefäßwiderstand bei links-rechts-Shunt -> im Verlauf rechts-links-Shunt
Ät.: VSD, ASD, PDA, etc. Endotheliale Dysfunktion, Thombozytenaktivierung Klassifizierung nach morphologischen Kriterien in der Lungenbiopsie Kl.: -Zyanose, reaktive Polyglobulie -Belastungsdyspnoe -Synkopen (niedriges HZV) -Arrhythmien -Hämoptysen (Lungeninfarkte) -zerebrovaskuläre Ereignisse (paradoxe Embolien) -Müdigkeit, Schwindel, Sehstörungen (Hyperviskosität) Th.: -Vermeidung von ACE-H., AT1-Bl. -Senkung der pulm. HTN -Aderlass bei symptomatischer Hyperviskosität -evtl. O2 -Gerinnungsprobleme, Thrombozytenaggregationshemmer und Antikoagulantien möglichst vermeiden -Endokarditisprophylaxe |
|
|
Fontan-Operation
|
Palliativoperation bei komplexen angeborenen Herzfehlern: Univentrikuläres Herz
Zentralvenöses Blut wird direkt, d.h. ohne Zwischenschaltung eines Pumpventriukels in den Lungenkreislauf geleitet. (Ein erhöhter Venendruck reicht als Kraft aus.) Probleme im Langzeitverlauf: -Stenosierungen im Anastomosenbereich, Verschlechterung der Ventrikelfunktion, etc. -Thromben in RA und Pulmonalis -Enterales Eiweißverlustsyndrom, lebensbedrohlich! -Rhyhtmusstörungen |
|
|
Ejektionsfraktion - Schweregradeinteilung
|
Normal >55%
Leichtgradig 45-55% Mittelgradig 30-45% Schwer <30% |
|
|
Frank-Starling-Mechanismus
Bowditch-Effekt |
Frank-Starling: Kraft-Spannung
Bowditch: Kraft - Frequenz |
|
|
Herzinsuffizienz Klinik
|
- Venenstauung (Hals, Zungengrund)
- Ödeme - Stauungsleber - Stauunggastritis - Stauungnieren Gemeinsame Symptome: - Nykturie (nächtliche Rückresorption von Ödemen) - Sympathikotone Überaktivität |
|
|
ABCD-Gruppen der Herzinsuffizienz (AHA)
|
A Pat. ohne Symptome mit Risikofaktoren für Herzinsuffizienz
B Keine Symptome aber Zeichen einer strukturellen Herzschädigung C Symptome bei strukturellen Schäden D Terminale Herzinsuffizienz |
|
|
Diastolische Relaxationsstörung
|
30-50% aller Patienten mit Herzinsuffizienz, häufiger bei Frauen
Anstieg LVEDP (>16mmHg) Assoziation mit art. Hypertonie (LVH), KHK, Diabetes mellitus E early filling (passiv) A active filling (Vorhofkontraktion) E/A <1 Rel. St. I (vermehrte aktive Füllung) E/A > 1, Pseudonormalisierung (kompensatorischer Anstieg des Drucks in LA) E/È korrliert mit PCWP |
|
|
Welche Medikamente können eine Herzinsuffizienz verschlechtern?
|
-NSAR
-Glukokortikoide -Glitazone -Verapamil, Dilthiazem -Alpha-Blocker -Interferon -einige Zytostatika -Trizyklische Antidepressiva, Lithium, Clozapin -Propofol -Beta-Mimetika |
|
|
Digitalis-Intoxikation
|
1. Brechreiz, Durchfall (Vagusaktivierung)
2. Farbensehen (Gelbstich) 3. Herz -Sinusbradykardie -AV-Knotentachykardie -AV-Blockierungen -Muldenförmige ST-Senkung Antidot: Digitalis-Antidot BM |
|
|
Wirkmechanismus und spezifische Nebenwirkungen verschiedener Diuretika
|
1. Thiazide: Blockierung des Na Cl -Kotransporters im frühdistalen Tubulus
NW: Stoffwechselstörungen (Erhöhung von Glukose, TG, Chol.) 2. Schleifendiuretika: Hemmung des Na K 2Cl Carriers im aufsteigenden Teil der Henle´schen Schleife NW: Hörverlust 3. Spironolacton: Hemmung der Na-Absorption und der der K-Sekretion im Sammelrohr NW: Gynäkomastie, Amenorrhoe, Stimmungsänderungen; Hyperkaliämie |
|
|
Herzglykoside
|
Hemmung der Na/K-ATPase
-> Erhöhung des transmembranösen Na-Gradienten -> Hemmung des Na/Ca Austauschers -> Anreicherung von Ca im Zellinneren Wirkungen: pos. inotrop, pos. bathymotrop, neg. chronotrop, neg. dromotrop Ca steigert, K und Mg vermindert Digitalisempfindlichkeit. Digoxin wird renal ausgeschieden, Digitoxin 60% renal, 40% enteral, bei Niereninsuffizienz Steigerung der enteralen Ausscheidung Indikationen: 1. Chronische systolische (!) Linksherzinsuffizienz 2. Tachyarrhyhtmie bei Vorhofflimmern KI: -WPW-Syndrom! -HOCM, frischer Myokardinfarkt -Bradykarde HRS -Kammertachkardie -Hyperkalzämie, Hypokaliämie |
|
|
Herztransplantation
|
Standardmethode: Orthotope HTX; Voraussetzungen: AB0-Blutgruppengleichheit, Fehlen von zytotoxischen AK im Empfängerserum gegen Spenderlymphozyten, Ähnlichkeit von Körpergröße und Gewicht
Immunsuppression: 3-fach Therapie: Ciclosporin A, Mycophenolat, Kortikoide; im Verlauf auch Imurek, Tacrolismus Akute Abstoßung: Schnelle Dickenzunahme der Hinterwand im Echo, verminderte systolische und diastolische Beweglichkeit, Relaxationsstörung Labor: aktivierte Lymphozyten und Lymphoblasten, Trop, CK Myokardbiopsie Th.: Kortison-Stoßtherapie; ggf. monoklonale AK gegen T-Lymphozyten Chronische Abstoßung: Manifestation besonders an den Koronargefäßen (Transplantatvaskulopathie)Keine A.p.-Beschwerden, da operative Denervation 10-Jahres-Überlebensrate bis 70% |
|
|
Ciclosporin A
|
Meilenstein in der Transplantationsmedizin
Der Wirkmechanismus auf das Immunsystem besteht in einer reversibelen Hemmung der Freisetzung des Interleukins – 2 aus aktivierten T - Helferzellen, welches die Aktivierung des zellulären Immunsystems steuert. NW: - arterielle Hypertonie - Nephrotoxizität - neurologische Beschwerden (Tremor, zerebrale Krampfanfälle) - Gingivahyperplasie - Leberfunktionsstörungen - Lymphome |
|
|
Mycophenolat
|
Transplantationsmedizin
Reversibler Hemmer der Inosinmonophosphat-Dehydrogenase (IMPDH), der daher die Synthese der Guanin-enthaltenen Nukleotide (Guanosin) hemmt. Da für die Proliferation von T- und B-Lymphozyten die De-novo-Synthese von Purinen unerlässlich ist, während andere Zellarten den Wiederverwertungsstoffwechsel benutzen können, wirkt MPA stärker zytostatisch auf Lymphozyten als auf andere Zellen, wodurch diese selektiv gehemmt werden. |
|
|
Tacrolismus
|
Tacrolimus greift in den Stoffwechsel von T-Zellen ein und hemmt deren Aktivität. Es bindet an den zytosolischen Rezeptor, einem sogenannten Immunophilin innerhalb der Zielzelle. Der Komplex aus Immunophilin und Tacrolimus lagert sich an die Serin-Threonin-Phosphatase Calcineurin. Calcineurin kann nun nicht mehr aktiviert werden. Dadurch werden vermindert Zytokine (z. B. IL-2, c-myc, IL-3, TNFα, IFN-γ) in den T-Zellen synthetisiert und daraus freigesetzt, wodurch die Reaktion des Immunsystems auf transplantierte Organe unterbunden wird.
|
|
|
5 Hauptformen der Kardiomyopathien
|
1. DCM Dilatative Kardiomyopathie
2. HCM Hypertrophische Kardiomyopathie mit oder ohne Obstruktion 3. RCM Restriktive Kardiomyopathie 4. ARVCM Arrhythmogene rechtsventrikuläre Kardiomyopathie 5. NKCM Nicht klassizifierbarte Kardiomyopathie |
|
|
Einteilung von Kardiomyopathien nach der Ätiologie
|
1. Inflammatorisch (chronische Myokarditis oder Autoimmunreaktion)
2. Ischämisch 3. Hypertensive Herzkrankheit 4. Valvuläre CMP 5. Metabolisch (endokrin, z.B. Diabetes; Speicherkrankheiten, Systemerkrankungen, muskuläre Dystrophien, neuromuskuläre Erkrankungen) 6. Toxisch (Alkohol, Medikamente, Cocain) 7. Peripartale CMP 8. Tako-Tsubo-CMP |
|
|
Tako-Tsubo-CMP
|
Broken Heart Syndrome
Reversible linksventrikuläre Dysfunktion mit apical ballooning. Ät.: Katecholamin-assoziierte mikrovaskuläre Dysfunktion, Koronarspasmen ST-Hebungen, Herzenzyme, blande Koronarien |
|
|
Dilatative CMP
|
DCM
Systolische und diastolische Herzinsuffizienz bei weitem LV Ät.: 1. Genetisch (verschiedene Erbgänge) 2. Umweltfaktoren -Virusinfektion mit Autoimmunreaktion -Chagas, Lyme -hypertensive, ischämische oder valvuläre Schädigung des Herzens -Alkohol -Systemerkrankungen (SLE, Sarkoidose) -Hämochromatose -peripartale DCM -Chemotherapie -Mangelernährung -Tachymyopathie |
|
|
Hypertrophe CMP
|
HNCM (Hypertrophische nichtobstruktive Kardiomyopathie)
HOCM (Hypertrophische obstruktive Kardiomyopathie) Häufige Ursache für plötzlichen Herztod bei jungen Sportlern Kl.: -Dyspnoe -Angina pectoris -höhergradige ventrikuläre Arrhythmien, Synkopen Echo: SAM Systolic Anterior Movement, säbelscheidenartiges Flussprofil Bei HNCM Belastung, ggf. zeigt sich dann erst die HOCM (Stress-Echo, RR-Abfall) Th.: -TASH: Transkoronare Ablation der Septumhypertrophie (mit Alkohol) -TSM (Transaortale subvalvuläre Myektomie) -Verpamil oder Beta-Blocker -KI: Digitalis, Sympathomimetika, Nitrate, ACE-Hemmer, AT-1-Blocker |
|
|
Restriktive CMP
|
RCM
Seltene Erkrankung mit diastolischer Relaxationsstörung 1. Myokardiale RCM-Formen: -idiopathisch -familiär -Sklerodermie -Amyloidose, Sarkoidose -Hämochromatose, M. Fabry 2. Endomyokardiale Formen: -Endomyokardfibrose (Afrika) -Hypereosinophilie (Löffler-Endokarditis) -Karzinoid (Hedinger-Syndrom) DD: Konstriktive Perikarditis |
|
|
Arrhythmogene rechtsventrikuläre Kardiomyopathie
|
ARVD
Häufige Ursache von plötzlichem Herztod bei jungen Männern (Sportler) Fibrolipomatöse Degeneration des rechtsventrikulären Myokards und rechtsventrikuläre Dilatation. M. Uhl evtl. Variante 40% pos. Familienanamnese Kl.: -Synkopen -Kammertachkadien -plötzlicher Herztod Im EKG in 10% der Fälle Epsilon-Welle Th.: -körperliche Schonung, kein Sport -Betablocker -ICD |
|
|
Non-Compaction-Kardiomyopathie
|
Angeborene Erkrankung des linksventrikulären Myokards, sporadisch oder familiär.
Typisch ist eine prominente Trabekularisierung der apikalen Hälfte des LV mit tiefen intertrabekulären Recessus (Persistenz des embryonalen Maschenwerks) Kl.: -Herzinsuffizienz -ventrikuläre Arrythmien -Thrombembolie |
|
|
Myokarditis
|
Ät.:
-infektiös (Viren: Parvo B 19, Coxsackie A und B, HHV6, Inluenza, Adeno, Echo, HIV, HCV; Bakterien: Staphylokokken, Enterokokken, Streptokokken, Borrelien, Diphtherie; Protozoen: Toxoplasmose, Chagas; Parasiten: Trichinen, Echinokokken) 2. Nicht-infektiös: RA, Kollagenosen, Vaskulitiden, idiopathische Fiedler-Myokarditis Virusmyokarditiden können infolge Kreuantigenität von viralen und myokardialen Strukturen zu Immunphänomenen führen. Kl.: Müdigkeit, Schwäche, Tachykardie, Rhythmusstörungen, -Herzinsuffizienz MRT: Fleckförmiges diffuses Late-Enhancement, Signalanhebung in T2-gewichteten Sequenzen Th.: Bei Nachweis von Virus-DNA ggf. antivirale Therapie, bei Nachweis von Auto-AK ggf. extrakorporale Immunadsorption, Kortikoide, Immunsuppressiva Prognose: 80% Ausheilung oder Persistenz harmloser Rhythmusstörungen; 15% Chronischer Verlauf mit DCM |
|
|
Aktue Perikarditis
|
Ät.:
1. Infektiöse Perikarditis (meist durch Viren: Coxsackie A und B, CMV, Parvo-B-19, Adeno, Echo, HIV; selten Bakterien, z.B. Tbc) 2. Immunologisch (SLE, Rheumatisches Fieber, Serumkrankheit, Postkardiomyotomie-Syndrom - Dressler-Syndrom) 3. Perikarditis epistenocardica 4. Perikarditis bei Urämie 5. Tumorperikarditis 6. Perikarditis nach Radiatio Kl.: 1. Trockene (fibrinöse) Perikarditis: Stechender Schmerz retrosternal, verstärkt im Liegen, bei tiefer Inspiration und beim Husten; Perikardreiben) 2. Feuchte (exsudative) Perikarditis: Beim Übergang von trockener P. zu feuchter P. werden die HT leiser, die Schmerzen und das Reibegeräusch lassen nach. Komplikation: Herzbeuteltamponade EKG: Typ Außenschichtschaden mit konkavbogigen ST-Hebungen, in der 2. Woche T-Negativierung Th.: -Behandlung des Grundleidens -Symptomatisch: NSAR, evtl. Steroide -ggf. Entlastungspunktion |
|
|
Herzbeuteltamponade
|
Behinderung der diastolischen Ventrikelfüllung, v.a. bei rascher Zunahme des Ergusses (kritische Ergussmenge 300-400ml)
Kl.: 1. Rückstau des Blutes vor dem rechten Herzen -prall gefüllte Venen (Jugularis, Zungengrund) -Kussmaul-Zeichen: Paradoxer inspiratorische RR-Anstieg in der Jugularvene -Leberkapselspannung mit Oberbauchschmerzen -evtl. Aszites 2. Low cardiac output -Körperliche Schwäche -RR-Abfall -Pulsus paradoxus: Inspiratorische Abnahme der RR-Amplitude -Tachykardie 3. Leise Herztöne |
|
|
Chronisch konstriktive Perikarditis
|
Narbige Folgezustände der akuten Perikarditis. Die Einengung des Herzens durch den narbig geschrumpften, z.T. mit Kalkspangen durchsetzten Herzbeutel führt zur Behinderung der diastolischen Ventrikelfüllung mit den Zeichen der Einflusstauung und bei längerem Bestehen zu einer Herzmuskelatrophie.
Th.: Dekortikation |
|
|
Risikokalkulatoren für kardiovaskuläre Ereignisse
|
PROCAM-Score: tödliche und nicht tödliche kardiovaskuläre Ereignisse
ESC-Score: Nur tödliche kardiovaskuläre Ereignisse Framingham-Risikokalkulator |
|
|
Nach welchen Erkrankungen muss man bei Pat. mit Myokardinfarkt <30 Jahre fahnden?
|
-Familiäre Lipidstoffwechselstörungen
-Antiphospholipidsyndrom -Hypothyreose -Vaskulitiden (Panarteriitis nodosa, Kawasaki-Syndrom, Takayasu-Arteriitis) -Koronaranomalien -Drogenanamnese (Cocain, Marihuana) -Hyperviskositätssyndrom (z.B. multiples Myelom) |
|
|
Bland-White-Garland-Snydrom
|
Ursprung einer Koronararterie aus der Pulmonalarterie
|
|
|
Akutes Koronarsyndrom
|
1. Instabile Angina pectoris (ohne Anstieg von Troponin)
2. NSTEMI (Instabile A.p. mit Anstieg von Troponin, ST-Senkungen, T-Veränderungen aber ohne ST-Hebung) 3. STEMI (ST-segment-elevation myocardial infarction; infarkttypisches EKG und ST-Hebung) |
|
|
Sonderformen der Angina pectoris
|
1. Prinzmetal-Angina (Variant-Angina): A.p. mit reversibler ST-Hebung ohne Anstieg von Troponin. Passagere Spasmen in Koronargefäßen. Erhöhtes Risiko für akutes Koronarsyndrom und Herzinfarkt
2. "Walking through Angina": A.p. zu Beginn einer Belastung, die bei weiterer Belastung verschwindet (Freisetzung vasodilatierender Medikamente) 3. "Angina nocturna": Nachts aus dem Schlaf heraus auftretende A.p. |
|
|
5 dramatische Ursachen des akuten Thoraxschmerzes ("big five")
|
1. ACS
2. Lungenembolie 3. Aortendissektion 4. Spannungspneumothorax 5. Boerhaave-Syndrom |
|
|
Typische und atypische Angina pectoris
|
Typische Angina pectoris (alle Kriterien erfüllt)
1. Retrosternale Beschwerden charakteristischer Ausprägung 2. Ausgelöst durch körperliche oder psychische Beschwerden 3. Rückgang der Beschwerden durch körperliche Ruhe und/oder Einnahme eins kurz wirksamen Nitrats Atypische A.p. 2 der Kriterien sind erfüllt. |
|
|
Belastungs-EKG
|
Horizontale, deszendierende oder sehr träge aszendierende ST-Senkungen
0,1mV in Extremitäten 0,2mV in BWA max. HF 220-Lebensalter submax. HF 200-Lebensalter |
|
|
Zielwerte LDL-Cholesterin
|
sehr hohes Risiko <70mg/dl
hohes Risiko < 100mg/dl mittleres Risiko <130mg/dl niedriges Risiko <160mg/dl |
|
|
Wirkmechanismen von antianginösen Medikamenten
|
1. Beta-Blocker: Verminderung von Nachlast und Herzfrequenz
2. Nitrate: Vorlastsenkung 3. Kalziumantagonisten (bei Unverträglichkeit von Beta-Blockern): Nachlastsenkung 4. Ivabradin (Procoralan): Ansenkung der Herzfrequenz |
|
|
Womit sind Drug-Eluting-Stents beschichtet?
|
-Sirolimus, Everolimus
-Paclitaxel |
|
|
Kontraindikationen für Adenosin
|
-WPW-Syndrom mit Vorhofflimmern
-Asthma bronchiale -AV-Block >1° -Sick Sinus Syndrom -QT-Verlängerung -Vorbehandlung mit Verapamil |
|
|
Schrittmacher Nomenklatur-Code
|
1. Stimulationsort
2. Detektionsort 3. Betriebsart 4. Programmierbarkeit 5. Antitachykardifunktion |
|
|
Schrittmachersyndrom
|
20% aller Patienten mit VVI-PM: Die Kammerstimulation führt zu retrograder Vorhoferregung und Vorhofkontraktion gegen die geschlossene AV-Klappe -> Verlust der Vorhofsystole und reflektorischer RR-Abfall mit Schwindelerscheinungen
|
|
|
3 Automatiezentren des Herzens
|
1. Sinusknoten, Ruhefrequenz 50-90/min
2. His-Bündel: Ersatzfrequenz 40-60/min 3. Purkinje-Fasern: Ersatzfrequenz 20-30/min |
|
|
Sick-Sinus-Syndrom
|
Degenerativer Schwund von Schrittmacherzellen oder sekundär (Myokarditis, Diphtherie, Borreliose, Medikamente)
Man unterscheidet: 1. Persistierende Sinusbradykardie (<40/min) mit inadäquatem Frequenanstieg unter körperlicher Belastung 2. SA-Block 3. Bradykardie-Tachykardie-Syndrom (typisch: lange präautomatische Pause beim Wechsel von schnellem VHF zu SR) |
|
|
Ventrikuläre ES
|
N VES N VES: Bigeminus
N VES VES N VES VES: Trigeminus N N VES N N VES: 2:1 VES N N N VES N N N VES: 3:1 VES VES VES: Couplet VES VES VES: Triplet VES VES VES VES... : Salve Lown-Klassifikation I Monomorphe VES (<30/h) II Monomorphe VES (>30/h) IIIa Polymorphe VES IIIb ventrikulärer Bigeminus IVa Couplets IVb Salven V R auf T-Phänomen (frühzeitiger Einfall einer VES während der T-Welle der vorangehenden Aktion) |
|
|
SA-Block
|
I Konstant verzögerte Erregungsleitung vom Sinusknoten zum rechten Vorhof, im Oberflächen-EKG nicht sichtbar
II Typ Wenckebach: Zunehmende Verkürzung der PP- Intervalle bis zum Ausfall, gleich bleibende PQ-Intervalle II Typ Mobitz: Plötzliche Pause, deren Dauer das Doppelte oder ganzzahlige Vielfache des vorausgegangenen PP-Intervalls ausmacht III Sinusarrest, Asystolie bis ein sekundäres oder tertiäres Schrittmacherzentrum einspringt |
|
|
Klassifikation art. Hypertonie
|
Optimal < 120mmHg <80mmHg
Normal 120-129/ 80-84mmHg Hoch-normal 130-139/ 85-89 mmHg Hypertonie Grad I 140-159/ 90-99mmHg Hypertonie Grad II 160-179/ 100-109mmHg Hypertonie Grad III >= 180/ >=110mmHg |
|
|
Formen der sekundären art. Hypertonie
|
1. Schlafapnoe-Syndrom
2. Renal (renoparenchymatös, renovaskulär) 3. Endokrin (Conn-Syndrom, Phäochromozytom, M. Cushing, Akromegalie, Hyperthyreose) 4. Gestationshypertonie 5. Andere (Aortenisthmusstenose, Aortensklerose, medikamentös, Drogen) |
|
|
Wie ist die maligne art. Hypertonie definiert?
|
-diastolischer Blutdruck >120-130mmHg
-aufgehobener Tag-Nacht-Rhythmus -Vaskuläre Schäden, insbeseondere Augenhintergrundveränderungen St. III-IV -Entwicklung einer Niereninsuffizienz |
|
|
Bei welchen Erkrankungen findet man Blutdruckdifferenzen zwischen beiden Armen?
|
1. Aortenbogensyndrom (Arteriosklerose, selten Vaskulitis, z.B. Takayasu-Arteriitis)
2. Stenose/ Verschluss der A. subcalvia 3. Aortenisthmusstenose mit Abgang der A. subclavia sinistra distal der Stenose 4. Aortendissektion |
|
|
Wie ist das Non-Dipping in der LZ-RR-Messung definiert und bei welchen Patienten kommt es vor?
|
Def.: Nächtliche RR-Absenkung <10mmHg
-LVH -Sekundäre Hypertonie (OSAS, Diabetes, Schwangerschaft, Schlaflosigkeit) |
|
|
Abklärung bei Pat. mit V.a. sekundäre art. Hypertonie
|
-Urin auf Katecholaminmetabolite
-Dexamethason-Kurztest -Hyperkaliämie (Conn Syndrom?) -Farbduplex der Nierenarterien -Nierenwerte -Schlafapnoe-Screening |
|
|
Wirkungen und Nebenwirkungen von ACE-Hemmern
|
-Senkung des peripheren Gefäßwiderstandes (AT-II-Verminderung)
-Volumenabnahme durch Drosselung der Aldosteron- und ADH-Produktion -Hemmung des Abbaus von Bradykinin (-> Vasodilatator, Hemmung des Gefäßremodelings, aber auch Reizhusten, angioneurotisches Ödem -Hemmung der aldosteroninduzierten Myokadfibrose -ggf. Hyperkaliämie (bei Kombination mit Kalium-sparenden Diuretika) KI: Nierenarterienstenose, Aortenstenose, HOCM |
|
|
Kalziumantagonisten
|
1. Verapamil-Typ, Diltiazem-Typ: wirken am Herz und an den Gefäßen, Klasse IV-Antiarrhythmika
2. Nifedipin-Typ (z.B. Amlodipin): Wirkung nur an den Gefäßen NW: -Reflextachykardie bei Nifedipin-Typ -Flush, Kopfschmerzen -Knöchelödeme |
|
|
Antihypertonika der Reserve
|
1. Alpha Blocker (Doxazosin - Diblocin, Urapidil - Ebrantil)
2. Zentral wirkende Sympatholytika: -Alpha-2-Agonisten (Clonidin - Catapresan; NW: Sedierung, Mundtrockenheit, Depression, Bradykardie, KI bei Sick Sinus oder AV-Block >1) -Moxonidin - Cynt -Methyldopa - Presinol; NW: AIHA, Lupus, Na und Wasser Retention, Gynäkomastie, psychische Störungen) 3. Arterioläre Vasodilatatoren -Dihydralazin - Nepresol (NW: reflektorische Tachykardie, medikamentöser Lupus) -Minoxidil - Lonolox (NW: reflektorische Tachykardie, Hypertrichose) 4. Reninhemmer (Aliskiren - Rasilez) |
|
|
Antihypertensiva in der Schwangerschaft
|
1. Dauertherapie:
-Methyldopa -Metoprolol -Dihydralazin 2. Notfalltherapie mit generalisierten Krämpfen -Magnesium 2-5g langsam i.v. oder Diazepam 5-10mg langsam i.v. -Dihydralazin oder Urapidil |
|
|
Schellong-Test
|
10 Minuten Liegen, 10 Minuten Stehen
Normale Reaktion: RR-Abfall systolisch <20mmHg/ diastolisch <10mmHg 1. Sympathikotone orthostatische Hypotonie: Abfall des syst. RR >20mmHg, Anstieg der Hf >=16/min 2. Asympathikotone orthostatische Hypotonie: RR-Abfall systol. >20mmHg, Hf gleichbleibend oder fallend 3. POTS (=posturales orthostatisches Tachykardiesyndrom): Anstieg Hf um >30/min oder auf >130/min ohne Hypotonie |
|
|
Schock - Definition und pathophysiologische Einteilung
|
Def.: Kritische Verminderung der Mikrozirkulation mit Hypoxie der Gewebe und metabolischen Störungen.
1. Hypovolämischer Schock 2. Kardiogener Schock (Kontraktionsschwäche, Volumenbelastung, Druckbelastung, Füllungsbehinderung, Herzrhyhtmusstörungen) 3. Versagen der peripheren Kreislaufsituation (septischer Schock, anaphlaktischer Schock, neurogener Schock) Gewebehypoxie -> Ansammlung saurer Metaboliten -> capillary leak Die präkapillären Gefäßabschnitte reagieren auf die Azidose empfindlicher als die postkalpillären -> Atonie der präkapillären Gefäße bei noch bestehender Konstriktion der postkapillären Gefäße -> Sludge-Phänomen der Erythrozyten, Mikrothromben |
|
|
Drotrecogin (Xigris)
|
Die Eigenschaften von Drotrecogin sind mit denen des endogenen humanen aktivierten Protein C vergleichbar. Das körpereigene Protein wirkt antithrombotisch, indem es die Faktoren Va und VIIIa blockiert. Über die Hemmung von PAI-1 fördert der Wirkstoff die Fibrinolyse. Aktiviertes Protein C unterdrückt zudem die Freisetzung der entzündungsfördernden Zytokine Interleukin-1 und TNF-α.
Indikation: Schwerer septischer Schock |
|
|
Karotis-Sinus-Syndrom
|
Überempfindlichkeit der Barorezeptoren im Bereich der Karotisgabel
2 Typen: 1. Kardioinhibitorischer Typ (90%): Vagusreizung führt zu Bradykardie oder Asystolie 2. Vasodepressorischer Typ (10%): RR-Abfall >50mmHg ohne wesentliche Bradykardie |
|
|
AV-Block II
|
II Typ Wenckebach: supra-His, gute Prognose, Vorhofersatzrhyhtmus
II Typ Mobitz: intra- und infra-His: schlechtere Prognose, Kammerersatzrhythmus, bei Symptomen PM-Indikation, Gefahr von Morgagni-Adams-Stokes-Anfällen. Keine Gabe von Atropin! (Gefahr des totalen AV-Blocks) |
|
|
Bifaszikulärer Block
|
z.B. RSB + LAH
ggf. PM-Indikation wenn -zusätzlich AV-Block I -HV verlängert -Schwindel oder Synkopen |
|
|
AVNRT
|
AV-Knoten-Reentry-Tachykardie
1. Gewöhnliche Form (95%, slow-fast): Eine SVES trifft auf den AV-Knoten zu einem Zeitpunkt, zu dem die schnelle Bahn noch refraktär ist, die langsame Bahn aber schon wieder erregbar. Die Erregung erreicht über die langsame Bahn das His-Bündel und kann die schnelle Bahn retrograd passieren. Jugularvenenpulsation, Hf 130-220/min, Pseudo-r´-Zacke in V, P-Welle im QRS-Komplex oder direkt danach 2. Ungewöhnliche Form (5%, fast-slow): Eine VES passiert die langsame Bahn retrograd und die schnelle Bahn anterograd. Retrograde P-Welle vor QRS, RP>PR. |
|
|
Differentialdiagnose einer SVT anhand der P-Welle in II/ III
|
P in der ST-Strecke, RP<PR: WPW oder atriale Tachykardie
Retrogrades P vor QRS, RP>PR: AVNRT (ungewöhnliche Form), atriale Tachykardie (AT), permanente junktionale Reentry-Tachykardie (PJRT) Antegrade P-Welle vor QRS: Sinustachykardie, atriale Tachykardie P-Welle im QRS-Komplex oder direkt danach: AVNRT (gewöhnliche Form) |
|
|
Welche Varianten der AVRT (atrioventrikulären Reentrytachykardie) unterscheidet man?
|
1. WPW-Syndrom
2. Mahaim Faser 3. Verborgene akzessorische Bahn 4. Permanente junktionale Reentrytachkardie |
|
|
WPW-Syndrom
|
Wolff-Parkinson-White-Syndrom
akzessorische, schnell leitende Bahn zwischen Vorhof und Kammer (Kent-Bündel, erregt Teile der Ventrikelmuskulatur vorzeitig - "Präexzitationssyndrom") PQ<0,12s, Delta-Welle, sekundäre Erregungsrückbildungs-Störungen, elektrisches Alternans in V2 - V4 bei Tachykardie. Sternalpositiver Typ: Delta-Welle in V1 positiv, QRS wie RSB -> Kent-Bündel linksseitig Sternalnegativer Typ: Delta-Welle in V1 negativ, QRS wie LSB -> Kent-Bündel rechtsseitig Tachykardie: Orthodrome Form: antegrad über den AV-Knoten, retrograd über die Bahn (schmale QRS-Komplexe) Antidrome Form: antegrad über die Bahn, retrograd über den AV-Knoten (breite QRS-Komplexe) Vorhofflimmern: FBI (Fast Broad Irregular) ->Verapamil, Digitalis und Adenosin sind kontraindiziert! (Gefahr Kammerflimmern, insbesondere Patienten mit kurzer Refraktärzeit der Bahn, prognostisch günstig: Verschwinden der Delta-Welle im Belastungs-EKG) Th.: Ajmalin, Flecainid, Propafenon, Kardioversion |
|
|
Verborgenes WPW-Syndrom
|
65% der Kent Bündel leiten sowohl antegrad als auch retrograd
5% leiten nur antegrad 30% leiten nur retrograd Bei ausschließlich retrograd leitenden Bahnen keine Delta-Welle, PQ normal! Hier Tachykardie nur orthodrom möglich, keine Gefährdung durch Vorhofflimmern. 30% aller WPW-Syndrome. Beim manifesten WPW können alle QRS-Komplexe eine Delta-Welle aufweisen (permanentes WPW) oder nur ein Teil (intermittierendes WPW) |
|
|
Mahaim-Syndrom
|
selten!
Akzessorische Bahn mit nur antegrader, langsamer Leitung Tachykardien antidrom PQ normal, keine Delta-Welle |
|
|
Permanente junktionale Reentry-Tachykardie
|
Akzessorische Bahn mit ausschließlich retrograden, langsamen Leitungseigenschaften.
Tachykardiefrequenz ist niedriger als bei normalen verborgenen Leitungsbahnen und wird vom Patienten auch wegen deren Permanenz oft nicht wahrgenommen. Kann zu Tachymyopathie führen. |
|
|
Fokale atriale Tachykardie
|
Vorkommen: Bei Gesunden, nach Herzoperationen, Herzerkrankungen, Digitalisüberdosierung
1. Unifokale FAT: Regelmäßige Tachykardie auf Vorhofebene mit monomorpher und unveränderter P-Wellen-Konfuguration, schmale QRS-Komplexe, Warming-up - Cooling-down 2. Multifokale FAT: Meist bei Herzerkrankungen. Mindestens 3 unterschiedliche P-Konfigurationen, häufig wechselnde PP- und PQ-Intervalle Bei Digitalisüberdosierung: AV-Bl II Tachymyopathie |
|
|
Vorhofflattern
|
Kreisende intraatriale Erregungsausbreitung in RA
Typ I (Common Type): Makro-Reentry gegen den Uhrzeigersinn, Flatterwellen in II, III, aVF negativ Typ II (reverse common type): Makro-Reentry im Uhrzeigersinn, Flatterwellen in II, III, aVF positiv Sägezahnmuster Atypisches Vorhofflattern (<3%): Mikroreentry, meist in RA Positive, nicht sägezahnförmige, abgegrenzte P-Wellen bei erhaltener Isoelektrischer in II, III; Vorhoffrequenz 280-380/min |
|
|
aberrierende Leitung
|
Eine Extrasystole kann auf die Kammer mit dem Bild eines Schenkelblocks übergeleitet werden. Unterschiedlich lange Refraktärzeiten beider Schenkel sind die Voraussetzung.
Bei Herzgesunden typischerweise RSB, bei organischer Herzerkrankung LSB. |
|
|
(nicht-) anhaltende VT
|
anhaltend >=30 Sekunden
|
|
|
Diagnostisches Vorgehen bei Breitkomplextachykardien
|
-capture-beat beweisend für VT (1-2 schmale vorzeitige QRS-Komplexe innerhalb einer Breitkomplextachykardie mit vorausgehender P-Welle
Kennzeichen einer VT: -Keine RS-Komplexe in allen BWA (Brugada 1) -RS-Abstand >110ms in BWA (Brugada 2) -Einfangschläge, Fusionsschläge -AV-Dissoziation -negative Konkordanz in den BWA -No man´s land weiterhin typisch für VT -Breite QRS > 140ms -Rabbit Ear in V1/V2 |
|
|
Differentialdiagnosen bei Breitkomplextachykardie
|
-VT
-SVT mit Schenkelblock (vorbestehend oder aberrierende Leitung) -antidrome WPW-Tachykardie -ventrikuläre Tachykardie bei Vorhofflimmern bei WPW |
|
|
Ionenkanalerkrankungen des Herzens, die zu Kammerflimmern führen können
|
1. Long-QT-Syndrom
2. Short-QT-Syndrom 3. Brugada-Syndrom |
|
|
Brugada-Syndrom
|
Mutation eines Natriumkanalgens, autosomal dominant
Coved-type- Hebung der ST-Strecke in V1, V2 ggf. erst im Ajmalin-Test sichtbar |
|
|
Leitsymptome des akuten Abdomens
|
1. Heftige Bauchschmerzen (umschrieben oder diffus)
2. Peritoneale Symptomatik (Abwehrspannung) 3. Störung der Darmperistaltik (Meteorismus, Übelkeit, Erbrechen) 4. Schlechter Allgemeinzustand, Kreislaufstörungen |
|
|
Nephritisches Syndrom
|
Volhard-Trias:
1. Hämaturie 2. Ödeme (Augenlider) 3. Hypertonus |
|
|
HAS-BLED-Score
|
Blutungsrisikoabschätzung vor Einleitung einer OAK
H Hypertonie A Abnormale Leber- oder Nierenfunktion S Schlaganfall B Blutungen L Labile INRs E Ältere (>65J) D NSAID, TAH oder Alkohol Maximal 9 Punkte |
|
|
Welches sind die häufigsten lebensmittelbedingten Durchfallerreger?
|
1. Campylobacter
2. Salmonellen |
|
|
Ätiologie der portalen Hypertonie
|
1. Prähepatischer Block: Pfortaderthrombose
2. Intrahepatischer Block: Leberzirrhose, etc. 3. Posthepatischer Block: Lebervenenthrombose = Budd-Chiari-Syndrom |
|
|
Impfempfehlungen für Erwachsene
|
1. Diphtherie, Tetanus, Pertussis alle 10 Jahre
2. Ab 60. LJ Pneumokokken, jährlich Influenza 3. HPV ab 18. LJ, Varizellen bei seronegativen Frauen mit Kinderwunsch 4. Masern, Röteln einmalige Auffrischung für nach 1970 geborene Personen 5. Polio als Reiseimpfung (nach insgesamt 5 Impfapplikationen lebenslanger Schutz) 6. Schwangerschaft: Influenza 7. Hepatitis A und B als Reisimpfung und für Risikopersonen 4. |
|
|
Tularämie
|
"Hasenpest"
Erreger: Francisella tularensis Ansteckung durch Aerosole, Verzehr von ungenügend erhitztem Fleisch, blutsaugende Vektoren (Zecken, Mücken), Jagen, Enthäuten, Schlachten, verunreinigtes Wasser Klinik: -Ulceroglanduläre Form, okkuloglanduläre Form, glanduläre Form -septischer Verlauf -Pneumonie -intestinaler Verlauf -abdominaler Verlauf mit Hepatosplenomegalie Letalität unbehandelt 30%, behandelt (Antibiotika) 5% Diagnose im Tierversuch, indirekter Erregernachweis schwierig, Kreuzreaktionen mit Typhus |
|
|
Wie kann man die Polymyalgia rheumatica von der Polymyositis/ Dermatomyositis differenzieren?
|
Polymyositis/ Dermatomyositis
-> CK! |
|
|
Rheumatoide Arthritis - Klinische Zeichen
|
-symmetrischer Beginn an den kleinen Gelenken (Finger), schmerzhafter Querdruck (Gaensslen-Zeichen), evtl. Gelenkergüsse, Muskelatrophie, Morgensteifigkeit
-Karpaltunnelsyndrom -Baker-Zyste -Rheumaknoten -Extraartikuläre Organveränderungen (Perikarditis, Myokarditis, Klappenläsionen, Pleuritis, Lungenfibrose, unspez. Leberenzymerhöhung, selten Glomerulonephritis, Keratokonjunktivitis sicca, Vaskulitis) -Sekundäres Sjögren-Syndrom Komplikationen: -Funktionsverlust und Fehlstellung von Gelenken: Schwanenhalsdeformität, Knopflochdeformität, ulnare Deviation, atlantoaxiale Subluxation -NW der antirheumatischen Therapie -Sekundäre Amyloidose -gehäuft maligne Erkrankungen und KHK |
|
|
Rheumatoide Arthritis - Klinische Zeichen
|
-symmetrischer Beginn an den kleinen Gelenken (Finger), schmerzhafter Querdruck (Gaensslen-Zeichen), evtl. Gelenkergüsse, Muskelatrophie, Morgensteifigkeit
-Karpaltunnelsyndrom -Baker-Zyste -Rheumaknoten -Extraartikuläre Organveränderungen (Perikarditis, Myokarditis, Klappenläsionen, Pleuritis, Lungenfibrose, unspez. Leberenzymerhöhung, selten Glomerulonephritis, Keratokonjunktivitis sicca, Vaskulitis) -Sekundäres Sjögren-Syndrom Komplikationen: -Funktionsverlust und Fehlstellung von Gelenken: Schwanenhalsdeformität, Knopflochdeformität, ulnare Deviation, atlantoaxiale Subluxation -NW der antirheumatischen Therapie -Sekundäre Amyloidose -gehäuft maligne Erkrankungen und KHK |
|
|
Positiver Rheumafaktor
|
5% bei Gesunden
40% im frühen Stadium einer RA, 80% im Spätstadium (seropositive RA) Sjögren-Syndrom Hepatitis C (durch Kryoglobulinbildung) |
|
|
Positiver Rheumafaktor
|
5% bei Gesunden
40% im frühen Stadium einer RA, 80% im Spätstadium (seropositive RA) Sjögren-Syndrom Hepatitis C (durch Kryoglobulinbildung) |
|
|
GERD - Stadien
|
Savary und Miller
0 Reflux ohne Schleimhautveränderungen I Isolierte Erosionen IA oberflächllich (rote Punkte) IB tiefer (rote Punkte, weißes Zentrum) II Longitudinal konfluierende Erosionen III Zirkulär konfluierende Erosionen IV Komplikationsstadium IVA mit entzündlichen Veränderungen IVB Irreversibles Narbenstadium Los Angeles A Eine oder mehrere Erosionen <5mm B Wie A aber >= 5mm C Erosionen erstrecken sich zwischen 2 oder mehr Kuppen, <= 75% der Zirkumferenz D Wie C aber >= 75% der Zirkumferenz betroffen |
|
|
Verlaufsformen monoklonale Gammopathie
|
1. Diagnose Plasmozytom:
>10% Plasmazellen im KM, monoklonales Protein im Serum/ Urin, CRAB-Kriterien (Ca, Krea, Hb, Knochen) a) progredient (90%) b) smolderung (10%) Ig>3g/dl, >10% PZ aber keine Endorganschäden 2. MGUS (Monoklonale Gammopathie unklarer Signifikanz): Konstant Ig<3g/dl, <10% PZ, keine Endorganschäden, keine Osteolysen. Risiko für die Entwicklung eines MM 1%/Jahr |
|
|
Gallenblasenpolyp
|
1. Cholesterolpolyp (häufigste Form): Gestielt, multipel, hypertrophierte mit Cholesterin beladene Zotten der Gallenblasenschleimhaut. Im Vergleich zum Lebergewebe echoreicher
2. Gallenblasenadenom: Ungestielt, genauso echogen wie das Lebergewebe, birgt ein geringes Risiko der malignen Entartung, deshalb bei rascher Größenzunahme oder >1cm CHE |
|
|
Lithogener Index
|
Cholesterin/ (Gallensäuren + Phospholipide)
wenn >= 1: Cholesterinübersättigung |
|
|
Indikationen zur CHE
|
1. symptomatische Cholezystolithiasis (akute Cholezystitis) -> frühelektiv innerhalb der ersten 72 Stunden
2. Porzellangallenblase (folge einer chronischen Cholezystitis): Entartungsrisiko 20% 3. Schnell wachsende GB-Polypen und Polypen >1cm |
|
|
Sekundäre biliäre Zirrhose
|
Komplikationen nach rezidivierendem Auftreten einer Cholangitis
|
|
|
Akute steinbedingte obstruktive Cholangitis
|
Charcot-Trias:
1. Fieber mit Schüttelfrost 2. Ikterus 3. Rechtsseitiger Oberbauchschmerz und Koliken mit Schmerzausstrahlung in den Rücken |
|
|
PSC
|
Primär sklerosierende Cholangitis:
Sklerosierende chronische Entzündung der extra- und intrahepatischen Gallengänge, bei 80% der Patienten mit Colitis ulcerosa assoziiert. Klinik: Ikterus, Juckreiz unklare Oberbauchbeschw. Ko.: Biliäre Zirrhose, Steatorrhoe, Mangel an fettlöslichen Vitaminen, Osteoporose, Cholelithiasis, Pankreasbeteiligung, CCC, KRK, rezidivierende bakterielle Cholangitiden Di.: ERCP Methode der Wahl: Perlschnurartige Gangunregelmäßigkeiten, divertikelartige Gangaussackungen, diffuse Gangrarefizierung Histologie: Periduktale Fibrose, kein spezifischer Befund! pANCA Th.: -UDCA senkt Risiko für CCC, verbessert AZ und Juckreiz -Bei Infektionen: Ceftriaxon -Bei Stenosen Ballon-Dilatation, Stents -Im Terminalstadium: Lebertransplantation -Bei Juckreiz: Cholestyramin |
|
|
PBC
|
Primär biliäre Zirrhose (auch: "vanishing bile duct disease"): Zirrhotisches Spätstadium einer chronischen nichteitrigen destruierenden Cholangitis; wahrscheinlich Autoimmunreaktion.
>90% Frauen, meist nach 40.LJ, erhöhtes Risiko bei Zöliakie Klinik: -Pruritus = Frühsymptom! -Müdigkeit -Hepatomegalie, Splenomegalie -Maldigestion -Xanthelasmen, dunkle Hauttönung (Melanin) -Assoziation mit Autoimmunhepatitis, CREST-Syndrom, Hashimoto. RA, Sjögren-Syndrom Diagnose: AMA (95% der Fälle), Cholestaseparameter, Hypercholesterinämie Di.: Juckreiz + Labor + Histologie, Bildgebung unauffällig. Th.: -UDCA bessert Ikterus und Prognose -Cholestyramin bessert Juckreiz, ggf. Antihistaminika -Fettarme Diät Bilirubin >6mg/dl Lebenserwartung <2Jahre -> Lebertransplantation anstreben! |
|
|
Hepatische Osteopathie
|
Unkonjugiertes Bilirubin beeinträchtigt die Osteoblastenfunktion und begünstigt damit die hepatische Osteopathie.
|
|
|
Welche Erkrankung ist typischerweise mit Lk-Schmerzen nach Alkoholgenuss assoziiert?
|
M. Hodgkin
|
|
|
Was ist ein Hiatus leucaemicus?
|
Fehlen der mittleren Entwicklungsstufen innerhalb der Granulopoese, typisch für AML.
|
|
|
Harnsteinarten
|
1. Kalziumoxalat (75%)
2. Struvite = Magnesium-Ammonium-Phosphat (=Infektsteine) (10%) 3. Uratsteine (5%) 4. Kalziumphosphatsteine (5%) |
|
|
Hirsutismus
|
1.Ovariell:
-Polyzystische Ovarien (Stein-Leventhal-Snydrom; in 45% gleichzeitig metabolisches Snydrom) 2. Adrenal: -Morbus Cushing -AGS (Virilisierung) -Adipositas, Diabetes mellitus -Typ II (bds. NNR-Hyperplasie) 3. Andere endokrine Ursachen: -Akromegalie (eher Hyertrichose) 4. Medikamentös (Androgene, Anabolika, Progesteron, Kortikoide, Phenytoin, Spironolacton,etc.) |
|
|
Gynäkomastie
|
1. Östrogenüberschuss
-Östrogen- oder HCG-bildende TU des Hodens oder der Nebennieren -paraneoplastisch (SCLC) -Leberinsuffizienz (verstärkte Östrogenkonversion aus Androstendion und Testosteron) 2. Androgenmangel -Hypogonadismus -Klinefelter Syndrom (XXY) -Hyperthyreose 3. Medikamentös -Östrogene, Antiandrogene -Spironolacton -Omeprazol 4. Marihuanakonsum (Phytoöstrogene) 5. Idiopathisch |
|
|
Polyurie
|
-Diabetes mellitus
-Diabetes insipidus -Hyperkalzämie -Hypokaliämie -Niereninsuffizienz, polyurische Phase des akuten Nierenversagens -Herzinsuffizienz -psychogene Polydipsie |
|
|
Ursachen einer Bewusstlosigkeit
|
1. Zerebrale Ursachen (SAB, Sinusvenenthrombose, Embolien, Insult, entzündl. Erkrankungen)
2. Endokrin (Diabetes -> Hypo- oder Hyperglykämie; Myxödemkoma, Thyreotoxische Krise; Addison-Krise, hypophysäres Koma) 3. Toxisch (exogen oder endogen - Urämie, hepatisches Koma) 4. Respiratorisch (CO2-Narkose) 5. Elektrolytengleisungen (Hyperkalzämie, Hyponatriämie) |
|
|
Abgeschwächte/ verlangsamte MER
|
-Polyneuropathie (Diabetes, Alkoholabusus, etc.)
-Hypokaliämie, (Hyponatriämie) -Hypothyreose -Polyradikulitis -funikuläre Myelose bei perniziöser Anämie |
|
|
Gesteigerte MER
|
-Hyperthyreose
-Hypokalzämie -vegetative Dystonie -neurolog. Erkrankungen (Parkinson, ED, Hirnödem) |
|
|
Hypothyreose
|
A 1:5000 Neugeborene
B erworben: meist Hashimoto-Thyreoiditis; iatrogen Kl.: -körperlicher und geistiger Leistungsabfall -Kälteempfindlichkeit -Haut trocken, kühl, teigig (Myxödem), blassgelb, schuppend -trockenes, brüchiges Haar -Obstipation -heisere Stimme -Myxödemherz (Bradykardie, Herzinsuffizienz) -Arteriosklerose -evtl. Myopathie mit CK-Erhöhung -Zyklusstörungen, Schwangerschaftskomplikationen -Beim älteren Menschen häufig oligosymptomatisch |
|
|
Myxödemkoma
|
1. Hypothermie
2. Hypoventilation mit Hypoxie/ Hyperkapnie 3. Bradykardie, Hypotonie 4. Myxödem Th.: Glukose, Glukokortikoide, Thyroxin (initial 100-200yg i.v.) |
|
|
Hypothyreose Diagnostik und Therapie
|
Di.: Latent (TSH erhöht, T3, T4 normal) oder manifest
TPO-AK (thyroidale Peroxidase) = MAK (mikrosomale AK) und TgAK bei Hashinmoto erhöht. Th.: Manifeste Hypothyreose: Dauersubstitution mit T4, einschleichend Ziel TSH bei jungen Pat. 0,5-2,0mU/l bei alten Pat. 4-6mU/l Latente Hypothyreose: Bei Pat. <70J T4 wegen des Risikos einer Früharteriosklerose und bei Frauen zur Vermeidung von Schwangerschaftproblemen/ Infertilität |
|
|
Hyperhidrosis
|
-endokrin: Menopause, Hyperthyreose, Karzinoid, Phäochromozytom
-Neoplasien (Nachtschweiß!) -Hypoglykämie -Dumping -Infektionen -Herzinfarkt, Lungenembolie, Schock |
|
|
Einteilung der Thrombozytopenien
|
1. Bildungsstörungen im Knochenmark
a) Knochenmarksschädigungen durch Medikamente, Strahlen, Infektionen, Chemikalien) b) Knochenmarksinfiltration c) Osteomyelofibrose d) Reifungsstörungen der Megakaryozyten (Mangel Vit B12, Folsäure) 2. Gesteigerter Umsatz a) DIC b) Immunthrombozytopenien (ITP v.a. bei Kindern nach Virusinfekten; HIT - Heparin-induzierten Thrombozytopenie) c) Hypersplenismus d) künstliche Herzklappen, extrakorporale Zirkulation e) Thrombotische Mikroangiopathie (TMA) |
|
|
Gewichtsverlust
|
-Unterernährung, Anorexia nervosa, Bulämie
-Dysphagie -Malgdigestion (chron. Pankreatitis, Magenresektion) -Malabsorption (CED, Zöliakie) -endokrin (Hyperthyreose, Diabetes mellitus, NNR-Insuffizienz, HVL-Insuffizienz) -chron. Infektionskrankheiten -Tumoren |
|
|
Akute Tonsillitis
|
1. Streptokokkenangina/ Scharlach
2. Infektiöse Mononukleose 3. Angina Plaut-Vincenti 4. Herpangina bei Coxsackie A-Virusinfektion 5. Akute HIV-Infektion 6. Diphtherie 7. Sexuell übertragbare Infektionen: Syphilis, Gonkokken 8. Agranulozytose |
|
|
Gelenkschmerzen
|
1. Anamnese: symmetrisch (RA) oder asymmetrisch, Monoarthritis (infektös), Oligo-/Polyarthritis; Stamm oder peripher, Zeitpunkt
2. Labor: RF, CCP, ANA, ASL-Titer, Borrelien-AK, Harnsäure, HLA-B27 |
|
|
Myopathie
|
1. Medikamenten-induziert
2. Alkoholmyopathie 3. Einschlusskörperchen-Myositis (seltene schmerzlose Muskelerkrankung mit distal betonten Paresen) 4. Polymyalgia rheumatica (Schmerzen und Steifigkeit im Schulter-/ Beckengürtel, 50% gleichzeitig Arteriitis temporalis, prompte Besserung nach Kortison) 5. Muskeldystrophie 6. Myasthenia gravis (Doppelbilder, Ptosis, belastungsabhängige Muskelschwäche bes. der Arme, Thymushyperplasie bei 65%, Auto-AK gegen postsynaptische Acetylcholinrezeptoren) 7. Lambert-Eaton-Syndrom (Auto-Ak gegen präsynaptische Ca-Kanäle; paraneoplastisch bei SCLC, myasthenieartige Schwäche der prox. Extremitätenmuskulatur, jedoch Besserung bei Belastung) 8. Infektiöse Myositis (Coxsackie, Trichinen) |
|
|
Sklerodermie
|
1. Progressive systemische Sklerodermie
2. Zrikumskripte Sklerodermie/ Morphaea (befällt nie die Hände, ohne Beteiligung innerer Organe) 3. Mischkollagenosen 4. Sklerodermieartige Krankheitsbilder durch chemische Noxen (Vinylchlorid, Siliziumdioxid) 5. Eosinophile Fasziitis (Shulman-Syndrom; Schwellung der proximalen Extremitäten ohne Hände und Füße, Eosinophilie im Blut und in der Hautbiopsie) 6. Acrodermatitis chronica atrophicans bei Lyme Borreliose 7. Nephrogene systemische Sklerose nach Gabe von Gadolinium-haltigen KM bei schwerer Niereninsuffizienz |
|
|
Fieber
|
1.Infektionskrankheiten (subfebril bei Endokarditis lenta, Tbc, Pyelonephritis)
2. Drug fever 3. Kollagenosen, Vaskulitiden, Erkrankungen des rheumatischen Formenkreises 4. Familiäres Mittelmeerfieber 5. Hyperthyreose 6. Tumoren (NHL, M. Hodgkin) 7. Gewebeabbau (Herzinfarkt, Lungeninfarkt, Pankreatitis) 8. Hämolyse (Sichelzellenanämie, Transfusionszwischenfall) |
|
|
undulierendes Fieber
|
unregelmäßiger, wellenförmiger Fieberverlauf, z.B. einige Tage subfebrile Temperaturen, dann wieder normale Temperaturen
-M. Bang (Brucella abortus) -Pel Ebstein Fieber (M. Hodgkin) -AIDS |
|
|
Roter Urin
|
Schnellteststreifen auf Blut positiv:
Mikroskopie: -keine Erythrozyten -> Myoglobinurie, Hämoglobinurie -Erythrozyten: -Eumorphe E. -> postrenale Blutung -Akathozyten und E.Zylinder -> renaler Ursprung Schnellteststreifen auf Blut negativ: umgekehrte Aldehydprobe: positiv -> Porphyrie negativ ->rote Rüben, Rifampicin |
|
|
Leukozyturie bei sterilem Urin
|
-Fluor
-antibiotisch anbehandelter HWI -Gonorrhoe oder nichtgonorrhöische Urethritis -Urogenitaltbc -Reiter Syndrom -Analgetikanephropathie |
|
|
Ödeme
|
1. Erhöhter hydrostatischer Druck in den Kapillaren: Niereninsuffizienz, Rechtsherzinsuffizienz, venöse Abflusstörung
2. Verminderter onkotischer Druck im Plasma (nephrotisches Syndrom, exsudative Enteropathie, Hungerödeme, Leberzirrhose) 3. Gesteigerte Permeabilitt der Kapillaren (akute postinfektiöse GN, Angioödem) 4. Verminderte Lymphdrainage (Lymphödem) 5. Arzneimittel (Kalziumantagonisten, Minoxidil, NSAR, Steroide, Östrogene, Antidepressiva, Glitazone) 6. Zyklische Ödeme (PMS) 7. Endokrin bedingte Ödeme (Myxödem - prätibial) 8. Sklerodermie |
|
|
DD Akutes Abdomen
|
1. Pankreatitis
2. Nieren- /Harnleiterkolik 3. Gallenkolik 4. Perforation (Ulcus ventr./duod., Sigmadivertikulitis, Gallenblase) 5. Mechanischer Ileus 6. Akute Appendizitis 7. Eingeklemmte Bauchwandhernie 8. Mesenteriale Ischämie 9. Gyn. Erkrankungen (Extrauteringravidität, stielgedrehte oder rupturierte Ovarialzyste) 10. Disseziertes Aortenaneurysma 11. Pseudoperitonitis: Praecoma diabeticum, Porphyrie, Addison-Krise) |
|
|
Pruritus
|
-allergische Hautreaktion
-Niereninsuffizienz -Darmparasiten -Polycytämia vera -Cholestase -Eisenmangel -PBC und PSC -trockene Haut -Diabetes mellitus -maligne Lymphome -psychogen |
|
|
Chronische Hepatitis
|
1. Virushepatitis
2. Autoimmunhepatitis 3. Toxische und medikamentöse Leberschäden 4. PBC 5. Hereditäre Stoffwechselerkrankungen: -Hämochromatose -M. Wilson -Alpha1-Antitrypsinmangel |
|
|
Autoimmunhepatitis
|
80% Frauen
-kontinuierlich erhöhte Transaminasen -frühzeitige Verminderung der Syntheseleistung -erhöhtes Gesamteiweiß und Gammaglobulin -Histologisch Bild einer chronisch aktiven Hepatitis -Nachweis typischer AK, Virusmarker negativ ANA, SMA bei Typ 1 (klassische lupoide AIH) LKM1 bei Typ 2 (selten, bes. Kinder) Überlappungssyndrome mit PBC oder PSC möglich Immunsuppressive Therapie mit Kortikoiden und Azathioprin -> relativ gute Prognose |
|
|
Primär biliäre Zirrhose
|
PBC
Zirrhotisches Spätstadium einer chronischen, nicht-eitrigen destruierenden Cholangitis unbekannter Ursache >90% Frauen, meist >40J. Kl: -Pruritus = Frühsymptome -Müdigkeit -Hepatomegalie, Splenomegalie -Maldigestion (verminderte Gallensäurenexkretion) -Xanthelasmen, dunkle Hauttönung Überlappungssyndrome mit AIH Labor: AMA >95% pos. ANA 50% IgM-Erhöhung GGT, AP, Bili Hypercholesterinämie Th. -UDCA verbessert Prognose -bei Juckreiz: Colestyramin, ggf. Antihistaminika -Lipase, fettarme Diät Bei Bili >6mg/dl Lebertransplantation anstreben |
|
|
Primär sklerosierende Cholangitis
|
PSC
Sklerosierende chronische Entzündung der extra- und intrahepatischen Gallengänge 80% mit Colitis ulcerosa assoziiert, 5% der Pat. mit CU haben PSC Kl: Pruritus, Ikterus, Oberbauchbeschwerden, Gewichtsverlust Ko.: Biliäre Zirrhose, 8% CCC, erhöhtes Risiko für kolorektale Karzinome Labor: pANCA ERCP: Perlschnurartige Gangunregelmäßigkeiten Th: UDCA verbessert Prognose ggf. Lebertransplantation |
|
|
Erbrechen
|
1. Gastrointestinale Erkrankungen:
-Gastroenteritis -Pankreatitis -Ulcus -Peritonitis -Ileus, Subileus, Stenosen -diabetische Gastroparese -Achalasie, Zenker-Divertikel 2. Schwere Schmerzen 3. Migraine 4. Erhöhter Hirndruck, Meningitis, Enzephalitis, Schädel-Hirn-Trauma 5. Morbus Meniére, Kinetosen 6. Intoxikation (Alkohol, Lebensmittelintoxikation, Digitalis, Zytostatika) 7. Urämie 8. Schwangerschaft 9. Ionisierende Strahlen 10. Psychogen |
|
|
Blutungsquellen des Ösophagus
|
1. Varizen
2. Mallory-Weiss-Syndrom (Schleimhauteinriss im Bereich der Kardia) 3. Boerhaave-Syndrom (Ösophagusruptur; Trias: Erbrechen, akute Thoraxschmerzen, Hautemphysem) |
|
|
Achalasie
|
Degeneration des Plexus myentericus (Auerbach) im unteren Ösophagus. Durch den Untergang inhibitorischer Neurone fehlt die schluckreflektorische Erschlaffung des unteren Ösophagussphincters.
1. Primäre A. 2. Sekundäre A. bei Kardiakarzinom, Chagas, paraneoplastisch Kl.: -Dysphagie -Regurgitation von Speisen -Völlegfühl retrosternal Ko.: Karzinomatöse Entartung Rö-Breischluck: Sektglasform Manometrie (hyper-, hypo- und amotile Form) Th..: -Nifedipin 30 Minuten vor d. Essen -Ballondilatation -Botulinum-Toxin in UÖS (schlechte LZ-Ergebnisse) -Operative oder laparoskopische extramuköse Myektomie des UÖS |
|
|
Diarrhoe
|
1. Infektiös
2. Antibiotika-assoziiert 3. Lebensmittelvergiftung 4. Medikamente 5. Maldigestion (Postgastrektomie, Gallensäureverlustsyndrom, exokrine Pankreasinsuffizienz) 6. Malabsorption (Sprue, Laktasemangel, M. Whipple) 7. CED 8. Karzinom 9. Mikroskopische Kolitis 10. Hormonell: Hyperthyroese, Karzinoid, Gastrinom, Vipom, Addison-Krise) 11. Autonome diabetische Neuropathie 12. Reizdarmsyndrom |
|
|
Obstipation
|
1. Funktionell (Faserarme Kost, mangelnde Flüssigkeitsaufnahme, mangelnde Bewegung)
2. Reizdarmsyndrom 3. Situativ (fieberhafte Erkrankungen, Reise, Schichtarbeit) 4. Medikamentös (trizyklische Antidepressiva, Anti-Parkinsonmittel, Opiate, Verapamil) 5. Hypokaliämie (Circulus vitiosus), Hyperkalzämie 6. organische Darmerkrankungen (Adenom, Karzinom, stenosierende Divertikulitis, Briden, M. Crohn, Analfissur, Abszess) 7. Neurogen (diabetische autonome Neuropathie, M. Parkinson, Multiple Sklerose, M. Hirschsprung) 8. Endokrin (Hypothyreose, Schwangerschaft) |
|
|
Thoraxschmerzen
|
1. kardial:
-Angina pectoris -Aortenvitium, HOCM -Perimyokarditis -Tako-Tsubo 2. Pleural/ pulmonal -Lungenembolie -Pleuritis -Pleurodynie (Coxsackie) -Pneumothorax 3. Mediastinum -Mediastinitis -Aortendissektion 4. Ösophagus -Refluxkrankheit -Motilitätsstörungen -Mallory Weiss -Boerhaave-Sydrom 5. Rippen, Wirbelsäule -vertebragene Thoraxschmerzen -Tietze-Syndrom -Herpes zoster 6. Abdominalerkrankungen -akute Pankreatitis -Gallenkolik -Roemheld Syndrom (geblähter Magen, reflektorisch A.p.) 7. Schmerzhafte Krisen bei Sichelzellanämie 8. Funktionelle Thoraxschmerzen (Da Costa Syndrom) |
|
|
Erythema nodosum
|
1. A-Streptokokken
2. Salmonellen, Shigellen, Yersinien 3. Mykobakterien 4. Kontrazeptiva, Schwangerschaft 5. Sarkoidose 6. CED 7. Malignome 8. Idiopathisch |
|
|
diffuser Ösophagusspasmus
|
Ätiologie unbekannt
Klinik: Anfallsweise heftige retrosternale Schmerzen, Dysphagie; kein erhöhtes Risiko für maligne Entartung Röntgen: Korkenzieherartig gewundener Ösophagus Manometrie: Normaler Ruhedruck im UÖS, normale schluckreflektorische Erschlaffung. Simultane, verlängerte Kontraktionen von erhöhter Amplitude, keine Propulsion. Th.: Kalziumantagonisten vor dem Essen, pneumatische Dilatation, ggf. Längsmyotomie |
|
|
Nussknackerösophagus
|
Klinik: Angina pectoris-artige Schmerzen, Dysphagie
Röntgen: Perlschnurartig verlaufende Kontraktionen Manometrie: Normaler Ruhedruck im UÖS, normale schluckreflektorische Erschlaffung. Propulsive, hypertone, verlängerte Kontraktionen |
|
|
Eiweißelektrophorese
|
1. Akute Entzündung: Alpha 1 und 2 erhöht
2. Chronische Entzündung: Gamma erhöht 3. Leberzirrhose: Albumin vermindert, Gamma erhöht 4. Nephrotisches Syndrom: Albumin vermindert, Alpha 1 und 2 und Beta erhöht |
|
|
EKG-Stadien Myokardinfarkt
|
1. Erstickungs-T, dann ST-Hebung in mindestens 2 zusammenhängenden Ableitungen, >=1mV in EA, >=2mV in BWA
2. Zwischenstadium: R-Reduktion, Pardee-Q, T-Negativierung 3. Alter Infarkt: Tiefes Q, R kann sich teilweise wieder aufbauen, T wieder normal |
|
|
Gallensteine
|
90% Cholesterinsteine (lithogener Index: Cholesterin/ Gallensäuren + Phospholipide)
10% Bilirubinsteine 60-80% der Steinträger sind asymptomatisch. |
|
|
Pfortaderthrombose
|
Ätiologie:
-Pankreatitis (Milzvenen- und Pfortaderthrombose) -Pankreas-Ca -Leberzirrhose Klinik: -Splenomegalie -normale Lebergröße und -funktion -Ösophagus-/ Magenfundusvarizen -fehlender Aszites The: Antikoagulation, evt. lokale Lyse durch TIPSS |
|
|
Aszites - Ätiologie
|
1. hepatogen (Leberzirrhose)
2. kardial (Rechtsherzinsuffizienz mit Stauungsleber) 3. maligne (Peritonealkarzinose) 4. infektiös (bakterielle Peritonitis) 5. pankreatogen (Pankreatitis) 6. traumatisch |
|
|
Aszites - Exsudat - Transsudat
|
Exsudat: Eiweiß >2,5g/dl, S/A AQ: <1,1g/dl, spez. Gewicht >0,016g/l)
a) entzündlich (Neutros > 250/yl) b) maligne (LDH >200U/l, Cholesterin >45mg/dl) Transsudat a) portale Hypertension b) Hypalbuminämie |
|
|
Manifestation der Postprimär-Tbc
|
80% pulmonal
20% extrapulmonal: -LK -Pleura -Urogenitaltrakt (Morgenurin nach Dursten) -Knochen, Gelenke -Darm, v.a. Ileozökalbereich, histologisch Epitheloidzellgranulome, endoskopisch wie Morbus Crohn |
|
|
Lungenmetastasen
|
Prostata-Ca
Kolon-Ca: bis linke Flexur über die Leber, ab linke Flexur auch dikrekt in die Lunge Mamma-Ca Nierenzell-Ca SD-Ca |
|
|
Welche Erkrankungen kann man mit tiefen Duodenalbiopsien nachweisen?
|
1. Sprue
2. M. Whipple 3. Lamblien |
|
|
Aerobilie
|
1. nach Papillotomie (ERCP)
2. bilidigestive Anastomose 3. biliodigestive Fistel 4. Steinperforation in den Darmtrakt 5. gasbildende Bakterien bei Cholezystitis oder Cholangitis |
|
|
Mirizzi-Syndrom
|
Kompression des Ductus hepatocholedochus durch ein im Gallenblasenhals oder D. cysticus impaktiertes Konkrement
Trias: 1. Cholezystolithiasis 2. Cholezystitis 3. biliäre Obstruktion |
|
|
Therapie Malaria
|
M. tert./ quart.: Cloroquin, bei M. tert. im Anschluss 14 Tage Primaquin
Unkomplizierte M. tropica: Atovaquon + Proguanil (Malarone) Mefloquin (Lariam) Artemether + Lumefantrin (Riamet) Komplizierte M. tropica: Chinin + Doxycyclin |
|
|
Normwert Retikulozyten
|
0,5-2,5% der Erythrozyten (50.000 bis 100.000/yl)
|
|
|
Direkter Coombs-Test
|
Nachweis von IgG, die an Erythrozyten haften durch Zugabe von Coombs-Serum (Ak gegen Gammaglobulin) -> Agglutination
|