![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
102 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Prototipo de anemia regenerativa |
Hemólisis o hemorragia aguda |
|
|
|
Prototipo anemia hiporregenerativa |
Aplasia medular, déficit de hierro o déficit de B12 o Ácido fólico |
|
|
|
Hallazgo en sangre periférica en déficit de G6PD |
Cuerpos de Heinz |
|
|
|
Causa más frecuente de macrocitosis |
Alcohol |
|
|
|
Cuándo sospechar en aplasia medular? |
Pancitopenia y disminución de reticulocitos |
|
|
|
Criterios diagnóstico para Aplasia medular |

Criterios Camitta |
Por biopsia medular |
|
|
Clínica de aplasia medular |
Síndrome anémico, infecciones de repetición, fenómenos hemorrágicos |
|
|
|
Tratamiento aplasia medular |
Trasplante alogénico de progenitores hematopoyéticos de donante familiar en menores de 50 años Globulina antitimocítica o antilinfocitaria, esteroides, ciclosporina y Factores de crecimiento hematopoyético y granulo-monocitario |
|
|
|
Definición de Mieloptisis |
Ocupación de médula ósea por alguna patología que distorsiona la médula provocando salida de células inmaduras en sangre periférica Reacción leucoeritroblastica + dacriocitos + esplenomegalia |
|
|
|
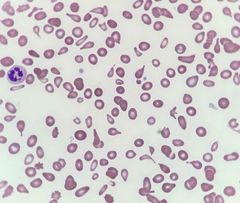
Frotis de sangre en Mieloptisis |
Dacriocitos/células en lagrima |
|
|
|
Etiología de la Mieloptisis |
Micrometastasis de Carcinoma en médula ósea, linfomas, leucemias, infiltración por vasculitis o granulomatosis y mielofibrosis primaria o secundaria |
|
|
|
Causa más frecuente de anemia |
Ferropenia |
|
|
|
Dónde ocurre la absorción de hierro? |
Duodeno y yeyuno proximal |
|
|
|
Tipo de anemia por déficit de hierro |
Anemia microcítica hipocrómica |
Dieta inadecuada, aclorhidria, cirugía gástrica, enfermedad celíaca, menstruaciones, sangrado crónico |
|
|
Clínica de anemia ferropénica |
Astenia, irritabilidad, palpitaciones, mareos, cefaleas, disnea, estomatitis angular, glositis, pagofagia(hielo), pica, coiloniquia, uñas en cuchara |
|
|
|
Diagnóstico de anemia ferropénica |
Disminución de VCM y HCM, aumenta de transferrina y disminución de saturación de transferrina, ferritina sérica baja(1er cambio detectado) y HbA2, ADE aumentado |
|
|
|
Diferencia entre anemia ferropénica y por trastornos crónicos |
Capacidad de fijación de hierro Elevada en ferropenia y normal en E. Crónica |
|
|
|
Tratamiento de anemia ferropénica |
Sal ferrosa 100-200 mg/día x 3-6 meses Embarazo dar 60 mg/día a partir de 2do trimestre hasta 3er mes postparto Dextrano-hierro IV o IM |
|
|
|
Valorar parámetros de ferropenia de mejoría con Tratamiento |
Reticulocitos suben a los 10 días de tratamiento Hemoglobina a los 2 meses Ferritina a los 3-6 meses |
|
|
|
Causa más frecuente de ferropenia |
Hombres: sangrado digestivo Mujer: pérdidas menstruales |
|
|
|
Anemia de enfermedad crónica o por mala utilización del hierro |
Anemia normocítica normocrómica(más común) Anemia microcítica hipocrómica |
|
|
|
Diagnóstico de anemia por enfermedad crónica |

Hiposideremia Disminución de concentración de transferrina Saturación de transferrina normal o disminuida Ferritina sérica incrementada
|
Tratamiento: de la enfermedad asociada NO dar hierro |
|
|
Dónde de se absorbe la vitamina B12 |
Íleon terminal |
|
|
|
Clínica anemia por déficit de B12 |
Glositis atrófica de Hunter, malabsorción, sx anémico y polineuropatías(alteración de sensibilidad propioceptiva y vibratoria) |
|
|
|
Etiología déficit de Vit B12 |
Veganos, déficit factor intrínseco, alteración íleon terminal, enfermedad celíaca, fármacos: biguanidas |
|
|
|
Diagnóstico de déficit por Vit B12 |
Concentración sérica de Cobalamina/B12 menor de 200 pg/dl, aumento de ácido metilmalonico > 270 y homocisteina en sangre >14, eliminación en orina de ácido metilmalonico |

|
|
|
Tratamiento déficit de Vit B12 |
VITAMINA B12 IM |
|
|
|
Causa más frecuente de malabsorción de Vit B12 |
Anemia perniciosa |
|
|
|
Clínica anemia perniciosa |
Aclorhidria + anemia por déficit de VitB12 |
|
|
|
Diagnóstico de anemia perniciosa |
Cobalamina en sangre, ácido metilmalonico, homocisteina, AC anticelula parietal gástrica y antifsctor intrínseco Prueba de Schilling: absorción de B12 al dar F. Intrínseco |
|
|
|
Causa más frecuente de anemia megaloblástica |
Déficit de folatos |
|
|
|
Sitio de absorción de ácido fólico |
Yeyuno |
|
|
|
Diagnóstico déficit de folatos |
Disminución de folato sérico <2 ng/ml Disminución de folato intraeritrocitario <100ng/ml |
|
|
|
Clasificación de anemia hemolítica |
Intracorpuscular o extracorpuscular Intravascular(aparato circulatorio) o extravascular(bazos) |
|
|
|
Clínica de anemia hemolítica |
Anemia, ictericia y esplenomegalia |
|
|
|
Esferocitosis hereditaria/Minkowski Chauffard |
Anemia hemolítica congénita más frecuente |
|
|
|
Clínica esferocitosis |
Ictericia, anemia, esplenomegalia, crisis hemolítica, crisis aplásica, colelitiasis, craneo en cepillo |
|
|
|
Diagnóstico de esferocitosis |
Elevacion LDH, bilirrubina indirecta, aumento de reticulocitos y policromarofilos, esferocitos aumento de CHCM y prueba de hemólisis osmótica |

|
|
|
Cuándo pensar en hemoglobinuria paroxística nocturna |
Pancitopenia + Anemia hemolítica + trombos de repetición en sitios inusuales |
|
|
|
Deficit de G6PD |
Ligada al X Anemia hemolítica enzimopática Cuerpos de Heinz Crisis hemolíticas por favismo, antipalúdicos, nitrofurantoina, sulfonamidas, analgésicos, vit K TX esplenectomia(hemólisis crónica) y ácido fólico |
|
|
|
Constitución de la hemoglobina normal |
4 cadenas de globulina y 4 núcleos hemo 97% Hemoglobina A1: 2 alfa y 2 Beta 2% Hemoglobina A2: 2 alfa y 2 delta 1% Hemoglobina F: 2 alfa y 2 gamma |
|
|
|
B-talasemias(A. Recesivo) |
Talasemia mayor/Cooley: disminución de Beta por lo que hay poca HbA1 y aumento de HbA2 y HbF Clínica: aumento EPO, hiperplasia medular(craneo en cepillo), pseudoquistes manos y pies, hipoxia tisular crónica, hemosiderosis secundaria, hepatoesplenomegalia, frotis: microcitosis e hipocromia. Dx electroforesis de hemoglobina demostrando menor Hb1A TX trasplante alogénico de precursores hematopoyéticos, esplenectomia, transfusiones, quelantes de hierro, hidroxiurea o butirato o azacitidina Talasemia menor: más frecuente, microcitosis, con hematíes incrementados, CHCM normal |

|
|
|
Anemia por células falciformes/drepanocitosis |
Cambio de ácido glutámico por valina Morfología de hoz/célula falciforme Anemia hemolítica + Crisis vasoclusivas: infartos renales, hueso, cerebro, pulmones, bazo(muerte por infecciones: salmonella) Dx electroforesis de Hemoglobina TX opiáceos, hidratación, vacunas, hidroxiurea, butirato y trasplantes de precursores hematopoyéticos |

|
|
|
Anemia hemolítica por Ac calientes |
Mujeres Etiologia: infecciones, leucemia linfática crónica B, LES, fármacos IgG Hemólisis crónica o crisis hemolíticas TX enfermedas de base + esteroide + esplenectomia, azatioprina o ciclofosfamida o rituximab |
|
|
|
Anemia hemolítica por Ac fríos |
Hemólisis intravascular IgM TX. Evitar el.frio, esteroides y rituximab NO esplenectomia |
|
|
|
Cuándo sospechar síndrome mielodisplásico |
Anemias refractarias a tratamiento en personas adultas > 70 años Anemia normo o macro, leucopenia(hiposegmentados/pseudoPelger, trombocitopenia MO: médula hiper hipercelular o hipocelular, micromegacariocitos, sideroblastos en anillo tinción de Pearl TX. Trasplante alogénico < 60 años o transfusiones, vit B6, ciclosporina, azacitidina |
|
|
|
Definición de poliglobulia |
HCT: mujer >48% hombre >52% Hb: mujer >16.5 hombre >18.5 Tx sangrías |
Neoplasia que produce poliglobulia con más frecuencia es hipernefroma |
|
|
Policitemia vera |
Neoplasia mieloproliferativa crónica caracterizada por aumento de eritrocitos Mutacion JAK2V617F cromosoma 9p Varones edad media Hb aumentada, VCM disminuido, rubicundez cutánea o mucosa, acúfenos, hiper viscosidad, hemorragia, trombosis, HTA, pérdida de peso, sudoración nocturna, hepatoesplenomegalia Muerte por trombosis |
TX sangrias + hidroxiurea + AAS o interferón o anagrelida |
|
|
Mielofibrosis primaria/agnogénica |
Megacariocitos proliferan en MO que mueren y provocan proliferación de fibroblastos: fibrosis Anemia e hipermetabolismo(sudoración nocturna, pérdida de peso,), visceromegalias secundaris HT portal, lesiones óseas Dx: reacción leucoeritroblastica en sangre periférica(células jóvenes en sangre), aspirado en seco con fibrosis medular TX trasplante alogénico de precursores hematopoyéticos en jóvenes, transfusiones, EPO, andrógenos, folatos y esplenectomia, hidroxiurea, talidomida |
|
|
|
Trombocitosis esencial |
Proliferación de megacariocitos plaquetaria Fenómenos hemorrágicos o trombosis, dolor urgente en manos, pies y dedos(eritromelalgia), infartos esplenicos Evoluciona a leucemia aguda Dx criterios OMS: proliferación megacariocitos en MO, plaquetas >450, presencia de JAK2V617F Tx AAS, hidroxiurea, interferón y anagrelida |
|
|
|
Leucemia mieloide crónica |
Proliferación serie mieloide 50-60 años.varones, cromosoma Filadelfia 9:22( BCR/ABL1) Clínica: hiper metabolismo, hepatoesplenomegalia, Sx anémico Sangre: glóbulos blancos en todas sus etapas, anemia.normo normo, trombocitopenia/trombocitosis, disminución FA y lactoferrina Medula hipercelular Dx traslocación t9:22 por FISH Tx trasplante alogénico, imatinib
|
Nota se considera leucemia aguda cuando hay más de 20% blastos en MO o 15% en sangre |
|
|
Leucemia linfática crónica |
Predominio linfocitos B Leucemia crónica más frecuente Clínica: asintomáticos, Sx anémico, síntomas B(fiebre, pérdida de peso y sudoración nocturna), hepatoesplenomegalia y adenopatías, infecciones de repetición NUNCA hay blastos en sangres solo hay linfocitos maduros CD5, CD19, CD20 +, linfocitosis, manchas de Gumprecht Deleción cromosoma 13q y 11q Tx ancianos: clorambucilo y jóvenes: fludarabina, ciclofosfamida y rituximab |
|
|
|
Tricoleucemia/células peludas |
Leucemia B con células con proyecciones citoplasmáticas Tinción fosfatasa ácida resistente a tartrato(FATR)+, CD25 + pancitopenia, esplenomegalia y adenopatías Dx biopsia Se asocia a vasculitis: panarteritis nodosa e infección por Legionella Tx: esplenectomia, interferón y pentostatina y cladribina |
|
|
|
Leucemia aguda |
20% o más de blastos en sangre periférica |
|
|
|
Leucemia más frecuente en pediátricos |
LLA más del 80% son de estirpe B |
|
|
|
Leucemias con cuerpos de Auer |
Mieloblásticas M2(maduración) y M3(promielocítica) |
|
|
|
Traslocaciones más común en LMA |
T8:21 |
|
|
|
Patrón característico en linfoma de Burkitt |
En cielo estrellado |
|
|
|
Traslocación más típica en leucemia mieloide crónica |
T9:22 puede evolucionar a LAL |
|
|
|
Clínica de leucemias agudas |
Sx anémico, neutropenia(infecciones), trombocitopenia, CID(M3), organomegalias(infiltración por blastos): hepatoespleno, adenopatías, dolor óseo, inf SNC, inf piel y encías(M4 y M5) Labs: citopenias, blastos periféricos Dx punción lumbar con más de 20% blastos aumento de lisozima o muramidasa en M4 y M5 y LDH y ácido úrico elevado |
|
|
|
Tratamiento leucemias agudas |
LAM: antraciclina y Ara-C, trasplante alogénico, y en M3 ácido transretinoico por CID y QT x 2 años LAL: vincristina, prednisona, L-asparaginasa, antraciclinas y ciclofosfamida, metrotexate, mercaptopurina y en T9:22 dar imatinib |
|
|
|
Células en linfoma de Hodgkin |
Células de Reed-sternberg(núcleo bilobulado) Células de Hodgkin(mononuclear) Célula lacunar |
|
|
|
Virus relacionado a linfoma de Hodgkin |
Virus Epstein Barr |
|
|
|
Linfoma de Hodgkin clásico |
Predominio linfocitíco: células reed-sternberg y células hodgkin, mejor.pronostico Esclerosis nodular: más común, bandas fibrosas, células lacunares, afecta mediastino y hay prurito Celularidad mixta: células reactivas inflamatoria(eosinofilos, neutrofilos) y Reed-sternberg o células de Hodgkin Depleción linfocitica: escasos linfocitos, hay células de Reed-sternberg y Hodgkin y pocos linfos pequeños |
|
|
|
Diseminación más frecuente de linfoma de Hodgkin |
Linfática |
|
|
|
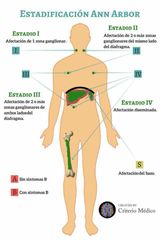
Clasificación de Ann-Arbor-Cotswolds |
Se añade A o B si hay ausencia o presencia de síntomas B |
|
|
|
Sintomas B |
Sudoración nocturna, fiebre tumoral y pérdida de peso inexplicable mayor de 10% en menos de 6 meses |
|
|
|
Masa Bulky |
Masa de más de 10 cm de diámetro en tórax |
|
|
|
Linfoma de Hodgkin linfocitíco nodular |
Crecimiento nodular con células L-H o en palomita de maiz con fenotipo B(CD45 y CD20) SIN CD15 o CD30 |
|
|
|
Clínica linfoma de Hodgkin |
Adenopatías periféricas, no dolorosas, Afectación mediastinica(esclerosis nodular), afectación esplacnica y abdominal(mixta), síntomas B, prurito, infecciones oportunistas |
Al ingerir alcohol se vuelven dolorosas las adenopatías |
|
|
Estudios complementarios en linfoma de Hodgkin |
Anemia de trastornos crónicos, leucocitosis con eosinofilia, linfopenia, VSG aumentada Anatomía patológica de ganglio |
|
|
|
Sindrome de Richter |
Transformación de leucemia linfocitica crónica a linfoma |
|
|
|
Tratamiento linfoma de Hodgkin |
Estadios limitados: 4 ciclos deABVD: adriamicina, bleomicina, vinblastina, dacarbacina + RT 20-30Gy Estadios avanzados: 6 ciclos de ABVD o BEACOPP: bloemicina, etoposido, doxorrubicina, ciclofosfamida, vincristina, procarbacina, prednisona |
|
|
|
Diátesis hemorrágica hereditaria más frecuente |
Enfermedad de Von Willebrand Anomalías cuantitativas o cualitativas del F VW, alteración en la.agregacion con ristocetina pero sin corregir con la administración de plasma normal |
|
|
|
Primer causa farmacológica de trombocitopenia |
Tiazidas |
|
|
|
Hemofilia A |
Ligada al X Leve 5-25% Moderada 5-1% Grave: inferior a 1% Clínica: hematomas tejido blando, hemartros,hemorragias internas, sangrado tras Cx Dx TTP alargado TP normal se comprueba al dosificar factor VIII TX factor VIII o acetato de desmopresina |
|
|
|
Cambios con la transfusión de 1 paquete eritrocitario 450 ml |
1 g/dl de Hb y 3% de HTO |
|
|
|
Principal etiología de Aplasia medular |
Idiopática |
|
|
|
Mejor parámetro bioquímico para detectar ferropenia |
Descenso de ferritina |
|
|
|
Prueba más fiable para diagnóstico de ferropenia |
Estudio directo de médula ósea |
|
|
|
Sangre periférica en déficit de B12 y B9 |
Hematíes de gran tamaño, neutrofilos hipersegmentados |
|
|
|
Causa más frecuente de macrocitosis sin anemia |
Alcoholismo |
|
|
|
Localizacion de los genes de las cadenas alfa |
Cromosoma 16 y 11 |
|
|
|
Única inmunoglobulina capaz de pasar la placenta |
IgG |
|
|
|
Causa más común de anemia en el mundo |
Anemia ferropénica |
|
|
|
Normal de saturacion de transferrina |
30-33% |
|
|
|
Riesgo de cáncer en anemia perniciosa |
5-10% adenocarcinoma gástrico |
|
|
|
Etiología leucemia aguda |
Radiación ionizantes, factor genético: anemia de Fanconi, ataxia-telangiectasia, neurofibromatosis, sx Down, benceno, cloranfenicol |
|
|
|
Linfoma de Hodgkin más frecuente |
Esclerosis nodular |
|
|
|
Causa más frecuente de trastorno hemorragico |
Trombocitopenia |
|
|
|
Tiempos de coagulación |

Tiempo de protrombina: coagulación extrinseca(III, VII, X, V) sirve para el control de coagulación oral. Tiempo de tromboplastina parcial activada: coagulacion intrínseca (XII,XI,X,IX, VIII, V): monitorizar tx con heparina no fraccionada Tiempo de trombina: mide fibrinogeno |
|
|
|
Factores dependientes de vitamina K |
II, VII, IX, X proteína C y S |
|
|
|
Trombocitopenia |
<100,000 plaquetas <50,000 plaquetas riesgo de sangrado postraumatico <20,000 sangrado espontaneo |
|
|
|
Causa mas frecuente de trombocitopenia por fármacos |
Tiazidas |
|
|
|
Purpura trombocitopenica inmunitaria |
Aguda: <6 meses, ant infeccion viral, niños Crónica:>6 meses, mujer joven, 65% se asocia a H. Pylori Son anticuerpos IgG Tx prednisona, esplenectomia, inmunosupresores: ciclofosfamida, azatioprina, ciclosporina, micofenolato,,gammaglobulina IV, danazol, plasmaferesis |
|
|
|
Purpura trombocitopenica trombotica |
Trombocitopenia con sangrado, anemia hemolitica microangiopatica, fiebre, afectacion neurologica transitoria y fluctuante y disfuncion renal Idiopatica: anticuerpos anti-ADAMTS13 Tx plasmaferesis |
|
|
|
Enfermedad de Bernard-Soulier |
A.Recesivo Ausencia de glucoproteina Ib y no puede adherirse al endotelio vascular |
|
|
|
Trombastenia/enfermedad de Glanzmann |
Fracaso en agregación de una plaqueta con otra por ausencia de complejo GPIIb/GPIIIa receptor para fibrinogeno |
|
|
|
Diátesis hemorragica hereditaria más frecuente |
Enfermedad de Von Willebrand Anomalía cuantitativa o cualitativa de factor vW Mal adhesión plaquetaria que corrige al agregar plasma Puede ser adquirida como en LES, gammapatias, hipernefroma o congenitas AD con disminución del factor VIII Clínica tiempo de sangría prolongado aparece tras Cx o trauma , sangrado ORL o equimosis Tx acetato de desmopresina o crioprecipitados |
|
|
|
Clasificación de Hemofilia |
Leve: 5-25% Moderado: 1-5% Severo: <1% |
|
|
|
Pacientes que no responden a heparina |
Deficit de antitrombina III |
|
|
|
Antídoto de heparina |
Sulfato de protamina |
|